Uncertainty vs Occurrence in QbD Risk Assessment
While extracting QTPP, CQA and CPP’s from the A-Vax case study, I noticed some authors used “uncertainty” instead of the popular “occurrence” for their QbD Risk Assessment.
What this means is that the Risk Priority Number (RPN) equation:
RPN = Severity x Occurrence
becomes
RPN = Severity x Uncertainty
(You can add detectability at the end. But here’s a reason why it’s unnecessary during the development phase.)
So is it Uncertainty or Occurrence in QbD Risk Assessment?
What is the Rationale for using Uncertainty?
At development phase, we usually don’t have enough data to determine the probability of occurrence. That is mainly why using uncertainty can make sense.
Uncertainty can help us identify where our knowledge gaps are. This helps us prioritize process parameters for the ensuing design space work.
Here are a few examples of how some QbD practitioners use Uncertainty in QbD Risk Assessments.
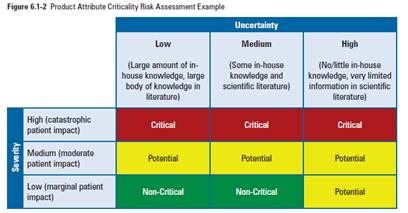
- Process Validation TR 60 from PDA (Process validation)
Here, PDA uses a simple decision algorithm for both Severity and Uncertainty.
Uncertainty:
- Low: Large amount of in-house knowledge, large body of knowledge in literature
- Medium: Some in-house knowledge and scientific literature
- High: No or little in-house knowledge, very limited information in scientific literature
Severity:
- Low: Marginal Patient Impact
- Med: Moderate Patient Impact
- High: Catastrophic Patient Impact
Here’s another example of using Uncertainty in QbD Risk Assessment.
- Application of the quality by design approach to the drug substance manufacturing process of an Fc fusion protein: Towards a global multi-step design space (Eon-duval et. al, Journal of Pharmaceutical Sciences Volume 101, Issue 10, pages 3604–3618, October 2012
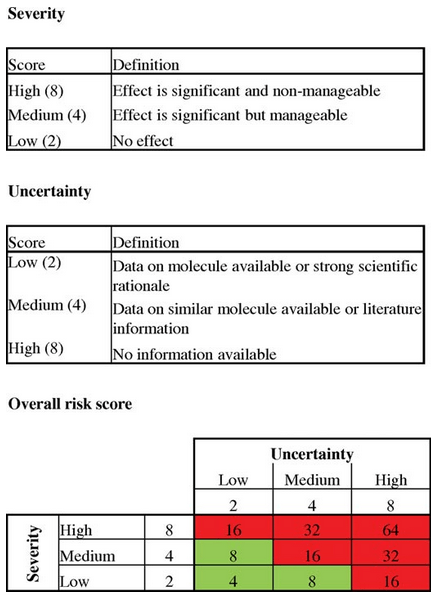
Here, Eon-duval et al used the following definitions for Severity and Uncertainty similar to PDA’s example:
Severity:
- Low: No Effect
- Medium: Effect is significant but manageable
- High: Effect is significant and non-manageable
Uncertainty:
- Low: Data on Molecule available or strong scientific rationale
- Medium: Data on similar Molecule available or literature information
- High: No information available
How can You Apply This?
After learning that some of our LeanQbD users were already using Uncertainty, we had to do something.
So we updated Lean QbD Software to be able to accommodate “Uncertainty” users.
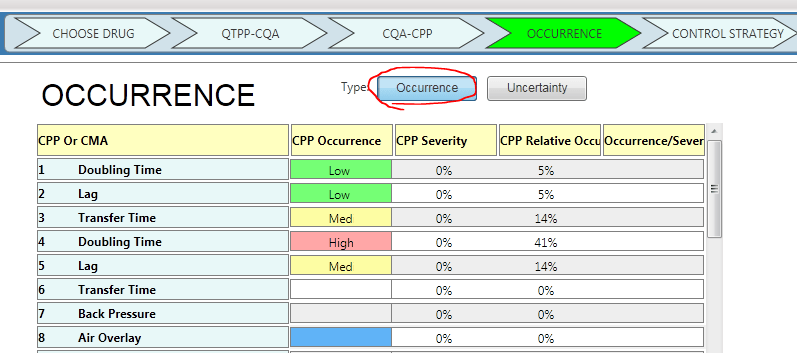
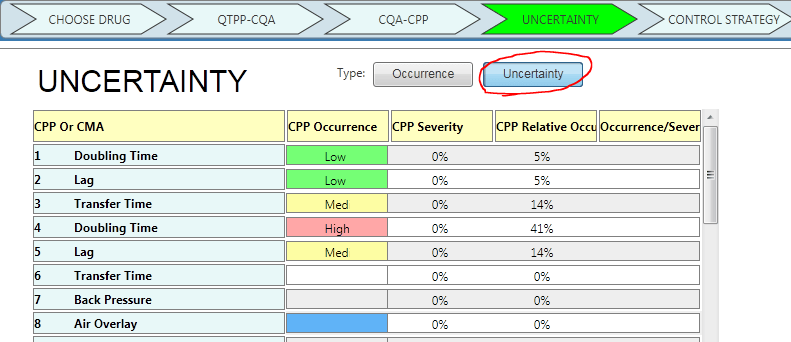
1. Use Occurrence or Uncertainty
Now, you have the option to select between Occurrence or Uncertainty. Mathematically, it doesn’t change the results.
You just ask a different question while facilitating the risk assessment. Instead of asking, “How likely is this process parameter to fail?” you are asking, “How little do we know about the effect of this process parameter?”
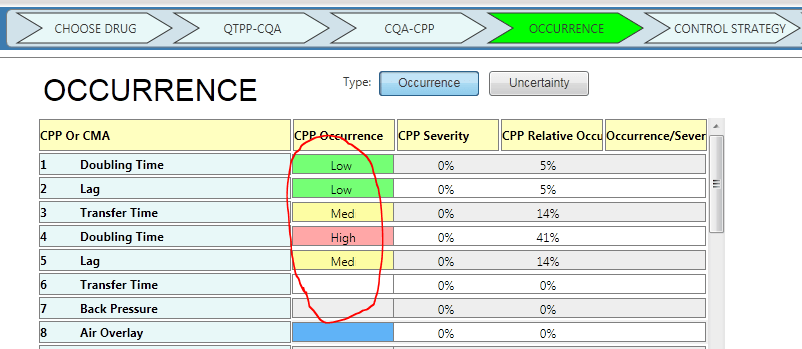
Below is a screenshot of how you can change your preference between occurrence and uncertainty. (Example from A-Vax template)
You can choose to use “Occurrence”
or “Uncertainty”
2. Simpler Rating Scale for Occurrence or Uncertainty: Low-Medium-High
Let’s not stop here. Let’s go one step further.
Remember the correlations between QTPP-CQA and CQA – CPP were “(none) – Low – Medium – High” while the rating scale for occurrence was 0-10? Some of you asked: Why do we need a 10 scale rating just for occurrence or uncertainty?”
You are absolutely right. At development stage, let’s keep the discussion simple and practical. Even PDA endorses the 3-level rating scale.
So we updated the rating scale to be consistent: (none) – Low-Medium-High.
Now you can ask, “How much (or little) knowledge do we have regarding this process parameter?”
If the answer is “very little,” then prioritize it for a design space study as many case studies suggest.
For our Current Users:
If you have been using the 1-10 scale, how will this affect your work? For now, we suggest that you revisit this section to update it.
If you’d like, we can automatically change the numbers to transform based on rating bands, i.e. 1-3: Low, 4-7: Med, 8-10: High.
But before we proceed, we wanted to confirm with you first. Would you like this feature or would you like to update it manually? Let us know!
We will continue to support both versions upon request.
Conclusion
Quality by Design is a goal. Its tools should be tailored to support the goal of understanding the design space.
You can use either “Uncertainty” or “Occurrence” in QbD Risk Assessments with confidence.
So my questions to you: For QbD Risk Assessment, which do you use?
Uncertainty or Occurrence?
You, Mehtap Saydam and 12 others like this
44 comments Jump to most recent comment
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Please comment on Which one you use (Occurrence or Uncertainty) and Why.
Delete 1 month ago
Amy Lachapelle
Amy
Amy Lachapelle
Founder of QbD Strategies
I agree with your view of the use of uncertainty at the R&D stage, especially when applied to drug attributes. That said, occurrence can be useful during process or analytical method R&D activities where you have basis for the probability of occurrence and need to include detectability of the occurrence.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Amy, good observation. Uncertainty as a start, use occurrence one you have enough info or data.
Delete 1 month ago
Sagar Panda
Sagar
Sagar Panda
Asst. Professor at Roland Institute of Pharmaceutical Sciences
For me its occurrence.
Like Reply privatelyDelete 1 month ago
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Sagar, thanks for sharing! I think more folks use occurrence because there are more examples. It shows that we can modify existing tools ie FMEA to fit to our own QbD needs.
Delete 1 month ago
P Venkat Bhaskar Rao
P Venkat
P Venkat Bhaskar Rao
Manager at Celltrion Chemical Research Institute
Dear Sun,
Thanks for sharing.
Considering, generic product development, where we generally do have information on molecule or a process, either from literature or by prior experience at the development phase, unlike as described.
Based on the available knowledge, it becomes easier to question OCCURANCE of an event rather than questioning our knowledge in terms of UNCERTAINITY.
Under, section “1. Use Occurrence or Uncertainty” when we compare the questions asked:
“How likely is this process parameter to fail?” Vs. “How little do we know about the effect of this process parameter?”
In case of the former question, we do have an idea about the answer in qualitative terms, and we optimize post risk assessment, to express in quantitative terms.
In case of the later question, it appears to be an open ended question.
In my work area, It was easier for me to correlate with OCCURANCE.
It most likely seems, that use of UNCERTAINITY, would very well suit in case of NCE or a new technology development, where the information from literature and experience are very limited.
Never the less, based on perception, an organization can take a call, on what to be used based on case by case basis, or use a single terminology, as the results do not differ.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Sagar Panda
Sagar
Sagar Panda
Asst. Professor at Roland Institute of Pharmaceutical Sciences
Dear Mr. Sun,
If we want to use Uncertainity instead of Occurrence then there should be sufficient back up information to support the use of the earlier one. Though QbD is being quite popularized now a days but people need to rationalize the meaning of UNCERTAININTY in different fields as the word itself creates confusion. Also the there should be some well defined set of terminologies and examples in the regulatory guidance for better understanding of the situation.However Occurrence is quite better term to use.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Dear P Venkat, I agree – for small molecules, occurrence is appropriate. Prof. Sagar, you are right that both Uncertainty and Occurrence need clear operating definitions to prevent a “rubber ruler” symptom.
Delete 1 month ago
Francesca Speroni
Francesca
Francesca Speroni
Project Leader at PTM Consulting srl
Dear Sun, first of all thank you for always sharing interesting topics!
Since I work both with R&D and operational I can use both terms: in fact, especially for assessments performed during early development stages, occurrence is very difficult to be estimated since knowledge and understanding are very poor… Differently, in later development stages, during transfer, or in operational environment,when knowledge increases, I do prefere the use of the occurrence since quantitative risk assmessent has to be more formal, and this term is known and understood all over. It can be easily communicated even to regulators since they are used to it…
Nevertheless, I agree that problems can income using both the terms and terminology and meaning have to be explained very well!
Unlike Reply privatelyDelete 1 month ago Pushpa Tiwari, Sun Kim like this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Francesca, great insight and summary from your experience of working with both R&D and Operations (I have the same privilege). So now my question: What are your DEFINITIONS for high-medium-low (or other) levels?
Delete 1 month ago
Francesca Speroni
Francesca
Francesca Speroni
Project Leader at PTM Consulting srl
Dear Sun, I’m sorry but, due to secrecy agreement, I cannot give you the exact definitions I usually use.. Nevertheless, talking about Uncertainty, I usually use three levels considering the knowledge and the “confidence” in this knowledge: definitions are generally qualitative in this exercise.
On the opposite when I use Occurrence, I try to be more quantitive and consider the estimated production in order to link the probability of a certain failure to the manufactured batches… In this way, it is easier to consider data coming from the manufacturing history in terms of deviations, non-ordinary maintenance, problems during manufacturing, etc.
Like (1) Reply privatelyDelete 1 month ago P Venkat Bhaskar Rao likes this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Francesca, thanks for sharing!
Delete 1 month ago
Dave Vasselin
Dave
Dave Vasselin
Associate Director at UCB Pharma
Its clearer to use uncertainty I feel when we discuss CQAs and their impact on the patient
For potential CPPs are you discussing uncertainty in determining severity/impact of a PP on a CQA or are you discussing the uncertainty regarding whether a PP will deviate from the defined set-point? For me then there is always some uncertainty in both
For both severity and occurrence may be its better to consider ‘error’ or ‘confidence’ with respect to the scoring. During the course of development the goal is to reduce the error as low as possible and then mitigate the residual risk
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
When dealing with process design considerations “neither” risk approach is adequate.
While risk tables and matrices are popular, quick, and easy to use tools for assessing levels of risk in a system there are many problems with them, and they are inappropriate for use in a process design setting–despite their wide acceptance. The pharmaceutical and supporting industries are taking a path fraught with “greater” risk using unproven methods of risk assessment at the process design stage. Cognitive ease will not solve this ensuing problem.
Now, I’m certain my comments will raise some ire with a few members of this discussion, and that’s fine with me. But, realize that no other industry that designs complex processes use “risk” methods as a basis for “selecting” candidate variables during process design. The only reason we do it is because of the momentum to do so placed upon us by regulatory agencies and prominent standards organizations to promulgate simple ways of conducting design, scale-up, and manufacturing applications in a quick and low-cost way. Designing pharmaceutical processes requires careful thought and consideration not simple methods that fast-track products to market.
There are a few pharmaceutical and life-science companies, (that hold their their methods close to their vest), who follow sound protocols for identifying CPPs at process design. These operations use proven life-cycle approaches built on an R&D construct and incremental process scaling framework to identify and verify the CPPs. Within this framework these companies understand their “true process design design risk” is “missing key process variables” (or, a Type II error), and work to minimize that error using robust design methodology.
Successful organizations recognize that Type II process design errors contribute to all other risks, e.g., patient, manufacturing, operational, and regulatory. Companies managing Type II design error recognize that understanding their process is key to managing the concomitant risks that are incalculable, but manageable within a controlled design and manufacturing schema. In the coming years these companies will become the industry leaders, and in fact some already have rightfully achieved that status.
Here are a few links to reference information that you may find useful:
http://www.spcpress.com/pdf/DJW230.pdfhttp://eight2late.wordpress.com/2009/07/01/cox%E2%80%99s-risk-matrix-theorem-and-its-implications-for-project-risk-management/
.
http://www.ncbi.nlm.nih.gov/pubmed/18419665
.
http://www.cox-associates.com/Coxbio.htm
.
http://academic.brooklyn.cuny.edu/economic/friedman/rateratingscales.htm
.
http://eight2late.wordpress.com/2009/08/09/cognitive-biases-as-project-meta-risks/
.
http://psiexp.ss.uci.edu/research/teaching/Tversky_Kahneman_1974.pdf
.
http://eight2late.wordpress.com/2009/10/06/on-the-limitations-of-scoring-methods-for-risk-analysis/
.
https://www.linkedin.com/groups/Risk-Matrices-are-they-all-1834592.S.98432679
Unlike Reply privatelyDelete 1 month ago Sun Kim, fereshteh barei like this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Ken, thanks for sharing references and insights. Surprise, I actually agree with you. That’s why I use the Lean QbD tool – that takes the features of C&E, QFD, and FMEA.
Our QbD industry calls it “risk assessment” but other industries call it “product development” (or drug/therapy development in our case). It’s just that we deal with patients’ lives directly so we are more attuned to “risk.”
Delete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Sun, we are not working in the QbD industry. QbD is a methodology for conducting product design and development. Right?
Yes I’m aware of how C&E, QFD, and FMEA work to provide a structured approach to product design and development–or, at least the I’m aware of the claim. C&E has been shown to be ineffective as a design tool for complex operations involving multiple factor interactions. QFD is not a risk assessment tool, it is a product design management tool. FMEA is the only claimed risk assessment tool in this tool box. FMEA is considered a QUALITATIVE risk management tool. There are limitations in the use of qualitative tools for risk management. Do we all understand the limitations before we begin using these tools in a new manner to select candidate CPPs?
For instance, there are many challenges using ordinal values for the purpose of selecting candidate CPPs via qualitative risk management methods, as cited by the authors previously provided. Even if SMEs support the selection process there are problems, because the fundamental process is flawed.
A selection of candidate CPPs should be done on the basis of a developmental “lifecycle” approach using structured experimental methods along the way to identify CPPs with greatest statistical significance. Secondary problems with qualitative risk assessment methods are related to the multiplication of ordinal values for the purpose of determining a risk index value. The mathematics of such an operation is invalid, as shown by D. Wheeler and others.
Yes, I know the automotive industry, who started this approach, does this all the time. But, if you were to do an assessment of historical recalls by the automotive industry you will find they are increasing in time, severity, and cost, not decreasing. Additionally, the “severity” of the defects managed using qualitative approaches do not appear to be managed well, follow the history of the NASA space shuttle disasters to get a sense of how well these methods of risk management handle high risk events.
Research to date on the effectiveness of risk indices, risk matrices, and the FMEA approach have not produced promising results, see my earlier links. This means these methods are not very effective at managing risk. The empirical results via the automotive, aeronautics, and space industries to name a few seem to empirically support this claim. It appears to me we are moving head-long in the same direction as the other industries while at the same time convincing ourselves we are doing the right thing for the right reasons!
If the methods you cite: C&E, QFD, and FMEA are effective in REDUCING Type II errors, (missing key CPPs), in process design applications, as you suggest, then please point us to the studies that support/prove this claim…
Many of the folks reviewing this discussion are trained in the sciences. At present there is broad SCIENTIFIC and MATHEMATICAL evidence that QUALITATIVE methods of risk assessment are limited at best and ineffective in most applications. Does anyone viewing my rant have sound SCIENTIFIC and/or MATHEMATICAL “evidence” that qualitative methods of risk management in fact yield “sound results and are effective” within the QbD framework? If so, please provide them in this discussion thread so that we may review them.
The only reason I presently see why we use these simple and less efficient methods of risk assessment and/or evaluation is due to what Daniel Kahneman calls COGNITIVE EASE. Cognitive ease does not adequately justify these methods even if every regulatory agency and standards organization endorses them.
In the end, the patient will suffer the greatest loss due to our cursory approaches to the product and process development paradigms we are building!
Like Reply privatelyDelete 1 month ago
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Ken, I like your passion. 🙂 We discussed the shortcomings of FMEA and Risk Assessments (please search or scroll down for relevant discussion threads.) And we discussed how we can overcome them.
There is no one silver bullet that can prevent type 2 errors. Risk Assessment is a precursor to Design Space studies.
You have to agree that – with QbD (despite imperfect) – we are in a better shape than 5 years ago.
We all have to start somewhere. We are not pursuing perfection but progress. 😉
Let’s learn, pivot and improve – as we go.
Delete 1 month ago
Inna Ben-Anat
Inna
Inna Ben-Anat
Director, Global QbD Strategy and Product Robustness at Teva Pharmaceuticals
Hi,
Very interesting discussion…
I think we are missing an important point here. There are several Risk Assessment stages throughout product life-cycle, even prior product submission to the agency. We will never end-up with just qualitative Risk Assessment tool. There is an initial Risk Assessment (and yes, it is mostly qualitative), that could be based on prior knowledge and experience (qualitative), in-house ‘know how’ (qualitative), historical data mining (quantitative), screening DOEs (quantitative) and more. Then there is a Risk Evaluation/Confirmation/Mitigation stage where we verify CPPs criticality and optimize true CPPs using DOE/modeling/simulation etc.-actual experimentation stage, quantitative of course. Robustness assessment usually follows using either simulation tools (such as Monte-Carlo) or actual Process Capability if there is enough data. Then the Final (for this stage of the life-cycle) Risk Assessment is conducted, where initially identified high risks could be reduced as a result of established control strategies/design space. Each stage of the lifecycle (scale-up, process design, continuous verification) will trigger revisiting previous risk assessment, initially revising using mostly qualitative methods and building control strategy using quantitative methods.
Unlike Reply privatelyDelete 1 month ago Andrei Zlota, Sun Kim and 1 other like this
Olivier MICHEL
Olivier
Olivier MICHEL
Senior Consultant chez ALTRAN AG
Ken, thanks for sharing links and prompting high level discussion. As a new comer in QbD technicalities I am not able to comment in detail. However as a born formulator and developper:
– I am not inclined to use the uncertainty as a value in risk ranking and risk matrixing in development; rudely said, the job here is to gain knowledge with facts and figures, not to express uncertainties? I remember the imprinted procedure and tips for recording experiment in our notebooks with the mention “refrain from expressing personal judgment”
– I am not inclined to use QbD techniques until a fair or basic understanding has been gained
– but agrees QbD as a whole is a good approach when combining , and fairly balancing, the scientific based x risk based x knowledge based means
er… managed to get a calculation in my post, rating sort of, not so bad for a maverick? 🙂
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Amy Lachapelle
Amy
Amy Lachapelle
Founder of QbD Strategies
I think that there are some very useful points being made in this discussion. First, as Ken said, QbD is a methodology for product development and second, as Inna brought up, the methodology used should be taken as a “life-cycle” approach meaning that the tools used as well as the CPPs and CQAs need to be continually monitored, evaluated and updated. The FDA recommends the use of a variety of different tools to start the life-cycle approach but these are not mandated. The life-cycle approach is a guideline toward a flexible, more effective approach to biopharmaceutical development. That said, given that this is not an exact methodology, it is critical that the teams working to implement this approach clearly define and document each term as it applies to the organization. That is, if using “uncertainty” vs “occurrence”, the team need to define and document what that means to the product or process and the rationale for assessing it.
I think QbD and life cycle management approaches provide a much needed strategy for the industry to begin implementing a more focused, success driven model that integrates different aspects of business and project management with the science.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Some very interesting and thought provoking recent comments. Thanks to all for their directed comments to me.
First, before I add a few comments to the mix I need to take care of some housekeeping. It appears that the LinkedIn editor bungled some of my earlier links that need to be straightened out. So, below are the two links that were previously concatenated together which I now separate allowing you to access properly:
Here is the link to Dr. Wheeler’s site explaining some of the problems using a Risk Priority Index value in doing FMEA risk assessment: (there are corollaries with present QbD methods). http://www.spcpress.com/pdf/DJW230.pdf
.
Here is a link explaining some of the problems using “heat maps” or “risk matrices” in a project management setting, based on the work of Dr. Tony Cox:
.
http://eight2late.wordpress.com/2009/07/01/cox%E2%80%99s-risk-matrix-theorem-and-its-implications-for-project-risk-management/
.
Next, I add a few new reference links supporting risk analysis, risk assessment, and risk planning:
.
* bible for risk analysis and management is Dr. Tony Cox’s reference as follows:
“Risk Analysis of Complex and Uncertain Systems,” Dr Louis Anthony Cox, Jr. Part of the International Series in Operations Research and Management Science, Springer Science and Business Media, LLC, c. 2009. See more details at the Amazon link below:
.
http://www.amazon.com/Analysis-Uncertain-International-Operations-Management/dp/0387890130/ref=sr_1_1?ie=UTF8&qid=1416532942&sr=8-1&keywords=Risk+Analysis+of+Complex+and+Uncertain+Systems
.
Here again is a link to Dr. Cox’s CV so that you can see this guy is serious about risk management practices and methods:
.
http://www.cox-associates.com/Coxbio.htm
.
An excellent book that covers the history of and uses of most risk management methods is Doug Hubbard’s book which also provides a clear detail on the issues with risk assessment methods used today.
“The Failure of Risk Management,” Douglas W. Hubbard, John Wiley and Sons, Inc., c. 2009. See more details at the Amazon link below:
.
http://www.amazon.com/Failure-Risk-Management-Why-Broken/dp/0470387955/ref=sr_1_1?s=books&ie=UTF8&qid=1416533258&sr=1-1&keywords=the+failure+of+risk+management
.
OK, I will close this post and start a fresh one containing some feedback to few comments above.
Like Reply privatelyDelete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
OK, let’s start with something simple first. I find when working with teams interested in conducting any type of risk assessment-analysis-management that they actually don’t know what the word “risk” means. I usually start the talk with what does the word “risk” mean to you. But, I know your time is short, so I’ll fill in the blanks.
“Risk is the chance, (actually probability), AND the magnitude of loss due to an undesirable event.”
So, we can say there is 85% chance of a tropical storm causing up to $10M of damage to a particular area. The nominal risk is calculated as (0.85)$10M, or $8.5M.
We can say that a particular medical product has a 5% chance of failure causing patient injury that would require about $150,000 of hospitalization and $5M of legal support. The nominal risk is: (0.05)$5.15M, or $257,500.
Sorry for the elaboration above, but I need it because we have a challenge. In the QbD approach we are asked to do something that is close to a paranormal experience. We are asked to look each parameter in a process and attempt to “estimate” the chance of each affecting a patient via the CQAs–“BEFORE WE EVEN UNDERSTAND HOW THE PROCESS ACTUALLY OPERATES.” Really? Wow!
I spent a few minutes looking over Sun Kim’s QbD software marvel and was immediately mesmerized! It’s slick, follows the QbD approach, sensible in application, uses lots of colors, and it’s exciting. I want to buy it right now!
But wait, I think back to my training as a process designer and developer. I immediately realize that’s not how I did the job of designing processes. Why? Well, you see I know that most chemical, pharmaceutical, biopharmaceutical, and vaccine processes contain two and possibly three factor interactions. I know that interactions are almost impossible to intuit using theory, common sense, and common experience via my limited senses. I look at the Cause and Effect Matrix built into Sun Kim’s cleaver piece of work and realize I could easily miss the subtle, but important interactions via this tool. Doing so would cause me to miss an opportunity to develop a truly efficient and robust process.
But, most important using “risk” as a basis for select candidate process control variables completely misses the point. It’s not “risk” I need to understand it “effect.” I need to know the effect of a process variable on the CQAs (e.g. responses). So, I ask different questions then those asked in a formal QbD exercise.
Oliver Michel and I share the same paradigm. Instead of sitting in front of a computer and playing with matrices and faux measures of risk I need to get out into the lab and get to work.
The first thing I will do is sit with the development scientists to gain some insight from them on the candidate variables likely to control the process. They may not be able to tell me the strength of the “effect” of each variable, but they can identify them from their experimental experience. Next I will organize a highly efficient set of Screening Experiments to gain a sense of which variables “might” actually control the process. Along the way I may even do a few characterization experiments.
When I have finished my pre-experimentation work I will return to the QbD Quality exercise and fill out the tables. But, this time the risk I use is the “risk” to process performance NOT the patient–because I have no clue about the causal chain of connection between a candidate CPP and the patient.
Along the way I will work hard as an experimenter to minimize the chance of claiming a truly unimportant variable is “significant” (Type I error), and most importantly of missing an important control variable (Type II error). For me the designer, experimenter, developer these two errors are the risks I need to manage in my work. This is where I need to focus my time and efforts. Less time on computers and more time in the lab is my goal!
I will close in the next posting..
Like (1) Reply privatelyDelete 1 month ago Alicia Tébar likes this
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
OK, now QbD has a place in the grand theme of things. QbD is actually a contract. It is an agreed upon way of presenting our findings to the regulatory agencies.
Over the past number of years industry and regulatory bodies have hammered out the details what one wants to see in submissions and what the other is capable of providing. QbD is the result of that negotiation. So, the quality and regulatory folks have some needs. They need to organize the results so that certain regulatory requirements are met. Without meeting these requirements there is little chance the new product submission will be acceptable by the regulatory reviewers.
Now whatever we do develop and scale up our processes we need to keep in mind the regulatory requirements. But, by the same token the designers and developers should not be hamstrung by arcane terms like “design space” in order to do their work. For example, I worked with a team a while back who felt they needed to display their established design space on paper as part of their submission, but they were having some problems. I asked them a simple question which they could not fully answer, and it was: “You have 5 CQAs and a variability term to display on a paper with two dimensions. That’s six responses or six dimensions to display in two dimensions, how can you possibly to do that in an intelligible way?”
We all need to get real. The case studies shown in Sun’s QbD workbook are ideal cases. Our applications will most likely be far more complicated. So, we need to be creative while realizing the folks who will be reviewing our work are NOT creative.
@Sun,
– It’s not passion you see from me. It’s concern!
– There is not one silver bullet to prevent Type II errors, because they are not preventable!
– Yes, I agree we are in better shape than 5 years ago, and even more confused!
– We should be pursuing perfection as an ideal, starting with the proper paradigms.
>>Note: you did not provide us with any links or connections on how to overcome the claimed shortcomings as you suggested in your posting above.
@Inna,
We missed no point. Our discussion focuses on the selection of CPPs up-front using an inefficient selection method, and I highlighted my experiences supporting such.
No amount of computer manipulation of data will replace actually getting out and doing the work that will allow the development team to understand the processes.
Yes you are right, it “is” all about a lifecycle approach to process development as I suggested in my earlier posts. But, if you start off in the wrong direction at the beginning by missing key interaction variables using an efficient “qualitative” single-factor method, then you will never regain the loss with more risk assessments down the line.
Finally, ask the question what happens when we miss an important process control variable during the initial stage of the development process–how do we mitigate that risk? Inna, can you give us all some of your insights how you have mitigated Type II errors in past work?
@Oliver,
I’m with you. I understand your challenges. You need to partner with a quality and regulatory associate so that you can work together to both develop a robust process and provide an accurate submission to the FDA that meets their criteria.
Much thanks to all for your time and patience.
Like (2) Reply privatelyDelete 1 month ago Alicia Tébar, Salvatore Buglione like this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Inna, Olivier, Amy, Ken, Thanks for adding value to the discussion. (we digressed from the topic of this thread – which is ok.)
Ken, thank you for bringing your energy to the discussion and for the links.
Many of us in this community read the books and papers and we reference them as well.
i.e. RPN fallacy (https://qbdworks.com/rpn-in-qbd-risk-assessment/) and why it’s necessary to separate severity and occurrence instead of just summarizing them by multiplication.
.
As Inna and Amy mentioned, “risk assessment” happens throughout the development and is updated with design space experiments (gaining process knowledge).
So everyone agrees that we should spend more time at the lab than messing with an excel spreadsheet or unnecessary-lengthy FMEA sessions.
(In fact, that’s why Lean QbD was born – to minimize non-value-add meetings so we can spend more time doing design space studies.)
We also discussed some time-saving techniques (https://qbdworks.com/2-uncommon-qbd-practices/) so we can focus more on process understanding.
FDA wants us to link QTPP-CQA-CPP/CMA. As drug manufacturers, we do need to understand how each process parameter will ultimately affect our patients.
Main reason? With limited time and resources, we must focus on more influential parameters. We have to prioritize the design space studies according to the importance of the parameters.
If interactions are important, it will show up in DOE’s and they will be updated on the risk assessment.
Yes, we also emphasize that case studies are ideal scenarios and should be taken with a grain of salt. Nevertheless, these working groups spent much time to co-author and share with the public some data points. We can still take what we need from these.
Do you realize all of us are trying to convey the same message? 🙂
“Do more design space experiments to increase process understanding”
It’s the community that evolves the practices of QbD so please keep challenging the status quo. This is a healthy discussion.
Delete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Thanks Sun for your follow-up. And yes, we are on the same page, but maybe not the same paragraph.
Remember, if one uses a risk table that scores 1/2 of an interaction pair low, then there is a “likelihood” that interaction may be missed during the experimental (AKA. design space) stage of development due to omitting the low scoring variable for further consideration. Thus, my concern with simple matrices for managing the flow of information at the early stage of process development.
If you say to me that you do not omit the low “risk” variables identified, then what is the purpose of evaluating all variables in a risk matrix?
These processes need to be internally consistent. Without consistency there is confusion!
Good work with the software tool, and excellent marketing approach! Kudos to you…
Like Reply privatelyDelete 1 month ago
Olivier MICHEL
Olivier
Olivier MICHEL
Senior Consultant chez ALTRAN AG
Sun, Kim et al, this is really a thrilling discussion. Maybe I can add a very recent experience from the field: we recently held a QbD presentation followed by a group exercise hosted by the Swiss Association for Quality. Topic was for naive and more experienced attendees (different industries, operational and quality managers,lean and opex …). Scope was to sort out and bring an action plan towards injectable manufacturing failures. Time was very short. Know what?, within the 4 constitued resolution groups, 2 jumped into an FMEA at once, 1 to an Ishigawa diagram; in my group we started with Ishigawa, but [hopefully?] switched before clock end to mapping the process; and this ended to be the fair start: as scientists or engineers we love jumping into tables and figures, but then we miss the essential part: know your process and your product or get to go knowing them better (and yes QbD tools and softwares can be of great help, but do not confuse tools with end)
Unlike Reply privatelyDelete 1 month ago Alicia Tébar, Sun Kim like this
Dave Vasselin
Dave
Dave Vasselin
Associate Director at UCB Pharma
I’d like to pose a small question regarding the point Ken made about Cause and Effect matrices. You said that in these matrices ‘risk’ is not assessed. I’m ‘uncertain’ about that statement. If you believe that there is a continuum of severity i.e. almost every parameter if stressed to infinite, perhaps ridiculous extremes will eventually cause a CQA failure, then all parameters would be defined as critical. Therefore when these matrices are completed there is either an implicit or explicit bracketing of a ‘reasonable manufacturing range’ which supposedly spans a plethora of potential failure modes either based on real manufacturing variance or perceived wisdom from production. Having made some kind of value judgement regarding a group of occurrences the matrix is completed on the basis of whether that particular parameter has any impact on a quality attribute.. Crudely both elements of risk are present, no?
For me the matrix is about what to do first and to what level of detail.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Matej Janovjak
Matej
Matej Janovjak
Unternehmensinhaber InSystea GmbH
possibly uncertainty may be considered within risk definitions e.g.
Severity I (Risk I) = Impact x Uncertainty (Bioprocess Study A –Mab)
Severity = Risk that attribute (+) impacts safety and/or efficacy
Impact = impact of attribute on CQA’s with subsequently impact on safety and/or efficacy
Uncertainty = uncertainty that attribute has expected impact
Regardless defintions, the key is and remain to understand causal relations between CPP’s,CMA’s and CQA’s in each process step as well as propagated accross the process.
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Dave, you hit on a key point. Use risk assessment as a prioritization tool (for design space work). Update with manufacturing-representative data.
Ken, good point on interactions hard to visualize on a 2-D matrix. Comforting thought is that when we iterate the PDCA cycles with “risk assessment” and “design space” studies, we usually find them.
In practicality, we do not have resources/time to test every variable, interaction, etc. As such, in DOE, we begin with Screening for main effects and then we look at interactions (sometimes together)
As you already know well, efficiency in finding the “critical” parameters is based on the principles of “effect sparsity” and “effect hierarchy” ( Wu, Hamada, 2000)
In industry, we balance efficacy and efficiency in our QbD methodology.
If not, scientist will say the usual “DOE takes too much time and resources.”
– which is a misunderstanding.
I do highly appreciate your thought-inducing discussion and this challenges us to improve.
Olivier, just like you, I also begin with process maps. This keeps the discussion focused and organized. (Hence, Lean QbD begins with a process map in a table format – It’s implicit so scientists can think at the levels of processes and unit operations.)
Ishikawa works well for focused Root Cause Analysis but is too broad for development so I don’t recommend it much in QbD. Start with Process Maps. Tools are not means but using the right tool definitely helps you run faster.
Matej, thanks for bringing the discussion back to the original scope: “Uncertainty or Occurrence.” I like how you broke down the definitions. Thanks!
Wonderful discussion!
Delete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Dave, if you feel a C&E matrix renamed a risk matrix does the job for you, then that’s great. In science we need to understand the potential error associated with our methods. Using a C&E matrix, PDCA, and production data to select candidate control variables we cannot evaluate our errors or test the method efficiency. This capability is lost in the initial part of the QbD process.
When developing new processes for a new product limitations in data availability make the variable selection process challenging. Bench-top and scaled research data are always useful for this task.
Sun, you said “good point on interactions hard to visualize on a 2-D matrix. Comforting thought is that when we iterate the PDCA cycles with “risk assessment” and “design space” studies, we usually find them.”
I recently had a wonderful opportunity to work with a team who followed the suggested QbD plan for process development. When they finished their development work they found there was a limited number of control variables and their processes did not perform very well.
When they called me in I looked over their work I noted they did everything correctly per QbD methodology, but something was definitely wrong. So, we went back to the drawing board. We reviewed the research data and noted there were no intermediate studies done to gain additional understanding and insight. Instead, there were the risk evaluations combined with data mining work of a “similar” process.
After a rigorous review of the research data we commissioned some initial studies and then went through the variable selection process. We ended up with 20 candidate variables screened to 12 variables using folded Res III designs. We then followed up that work with Res IV designs using center points to test for curvature in the model.
With the Res IV designs we were able to use sparsity of effects and heredity to elicit the significant interactions. In the end we followed a sequential experimental algorithm similar to that used for ages in other industries! The team was able to develop a predictive model with a high adjusted r-squared.
Next, I carried the team through a “Robust Design” effort using Variation Transmission Analysis to set the targets and tolerances for the identified CPPs. The complete development effort took two weeks longer than their original QbD work, but yielded a workable process operating a 4.8 sigma level of quality performance. Further tuning using in process EVOP brought the final performance to a 5.2 sigma level performance.
When using Risk Matrices/PDCA/C&E/In-house Know How/Data Mining etc. to SELECT candidate control variables there is a good chance of missing critical variables. But, that is not the real issue. The real issue is you will not know you missed them! Using these procedures for selection there is no “internal” way of testing for Type II errors as I suggested earlier. Given this lack of internal error control we cannot call the process of selecting candidate variables a “scientific” process.
No amount of word choices will change the lack of internal error control or lack of efficiency resulting from the use these methods. When developing pharmaceutical processes we should be using sound science as a basis. If we start our development work on shaky ground, then there is no way to recover even if we use the most sophisticated methods ever devised.
Again, I ask the experienced members of this discussion to provide us with the definitive research that shows the methods advocated to “select” candidate control variables for experimentation are both efficient and provide internal error control. Please provide full peer reviewed journal information and links if possible. Thanks much in advance.
Like Reply privatelyDelete 1 month ago
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Ken, thanks for your followup.
QbD is just a drug development framework. A framework for scientists and regulators to work together. It is not a problem solving tool or a DOE. ICH guidelines suggest but do not prescribe tools.
As for your experience with the team who claimed QbD but did not do the due diligence (” there were no intermediate studies done to gain additional understanding and insight”) … seems to have not understood QbD.
Just like six sigma or design controls, QbD results will vary on how it is implemented. Your expertise in problem solving, doe, evop saved the day by solving an isolated issue. No doubt these tools and methodologies are invaluable and should be part of the QbD tool set. Many of us here use them regularly.
Now when we try to communicate 1000’s of isolated incidents, studies, data, etc. to the regulatory agencies when we apply for our IND or BLA, it may not be easy.
Hence a summary, such as risk assessment acts as a guide for the reviewers. Furthermore, this summary also helps scientists prioritize their process characterization studies in chewable chunks.
To summarize:
QbD is a development framework, (not a problem solving tool).
To select the “critical” process parameters, (this goes beyond CTQ’s) there are many ways – ICH guideline coauthors preferred to use “risk assessment.”
Delete 1 month ago
fereshteh barei
fereshteh
fereshteh barei
Strategy advisor, Business analyst,Health and pharmaceutical industry
Excellent discussion, this gives me the idea of writting a new article.Thanks for sharing
Like Reply privatelyDelete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Sun, thanks back to you for your follow-up and thoughts above.
I’m impressed with your ongoing work organizing the process of translating patient needs and deploying them into the manufacturing process design effort via a software tool. In looking at your QbD tool, it carries protocol structures similar to Quality Function Deployment (QFD), but with some differences. I have to admit I needed a bit of time to look your tool over before I could provide cogent observations and/or questions via this forum.
In my spare time I reviewed your site reference info and followed through your QbD tool demo multiple times to gain understanding of its use. I enjoyed reading the section on your site entitled, “What Experts Do: RPN for QbD Risk Assessment” which allowed me to understand your thinking on the use of RPNs for assessing risk using ordinal values. I also reviewed the case studies provided via site links to assess what various folks were presenting at conferences on the application of QbD methodology.
Finally, I had a chance to read your section discussing the conceptual underpinnings of Shigeo Shingo’s SMED approach in combination with the Japanese concept of “Nemawashi” in preparing needed QbD information prior to any meeting structures. I’ve used similar methods in the past to better manage meeting time and was glad to see others have similar approaches using the same paradigms.
So before I comment on the “risk-based” approach for drug development and process design facilitated by your QbD tool, I would like to address a few recent comments you provided me in reference to my encounter with a company who followed QbD methodologty as illustrated in case studies and other literature–with limited success.
>>Your comment: “[QbD is] A framework for scientists and regulators to work together. It is not a problem solving tool or a DOE.”
Response: Yes Sun, the difference between problem solving and designing processes is clear to me. I have separate methods for doing each. My example above clearly explained I was supporting a process “design” effort.
>>Your comment: “As for your experience with the team who claimed QbD, but did not do the due diligence…”
Response: This team had a vast amount of experience in developing their product base, and referred to “prior knowledge” as a basis for moving through the QbD approach. They did not do intermediate studies, because they believed the methods provided via QbD would identify their areas of limited understanding–which it did not. So, they did their best with the knowledge they had available to them…
>>Your comment: “Now when we try to communicate 1000’s of isolated incidents, studies, data, etc. to the regulatory agencies when we apply for our IND or BLA, it may not be easy…”
Response: Yes, there is a large amount of variability in the application of QbD methods even though it has been in development for more than 8 years. Other industries did not take so long to find a structured protocol for developing process understanding and control. I still wonder why the pharma industry is so slow to achieve a consolidated approach to product and process design.
>>Your comment: “To summarize: QbD is a development framework, (not a problem solving tool).”
Response: Again, that is clear for me having done both problem solving and process design work for almost 30 years….
>>Your comment: To select the “critical” process parameters, (this goes beyond CTQ’s) there are many ways…”
Response: You see Sun, this is where we have a challenge. No one “selects” critical process parameters outright–not even using the Six Sigma approach. I understand the chain of relationship between Customer Needs to (CTQs or QTPPs) to CQA to CPPs. But, at the end of the chain I “select” POTENTIAL control variables to evaluate in a structured set of statistically designed experiments NOT CPPs.
See my next post for key questions on the QbD process.
Like Reply privatelyDelete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Sun,
What follows are a some comments in reference to your question of Occurance vs. Uncertainty, my observations from reviewing QbD case studies, a reiteration of the concepts of risk assessment from a subjective perspective, and finally a review of your QbD software tool.
Occurance vs. Uncertainty:
* As suggested by Sagar Panda and Francesca Speroni and others, there are no clear and definitive operational definitions for either words, “Occurance” or “Uncertainty” in the context of their use in the QbD approach. As such, they are used in any way necessary to achieve the purpose of demonstrating the operation of conducting a risk assessment, even if that is not the true purpose of the exercise.
* In determining the conditions for conducting a manufacturing process design, the utility of knowing the occurrence or uncertainty of an unknown quantity provides little utility for me…
Review of QbD Case Studies:
* A review of the QbD case studies provided via links on your site were of great interest to me. I had originally expected to see clear examples of how risk management methods were used to guide the process design effort through various stage of development. However, that is not what I observed. Instead, I observed a mix of confusing approaches to process development that did not seem to follow a coherent structure.
* On occasion, I observed the use of “risk matrices” that were implemented in non-standard ways. In reviewing these claimed risk tools it was difficult to understand how they were used to do anything other than to support selecting process variables that were identified from “prior knowledge or understanding.”
* In some of the presentations it was clear the development team understood how to properly use data mining and Neural Net methods to extract preliminary understanding from closely associated process data. But, these methods are not considered risk-based.
* I was taken back with some of the presentations that attempted to illustrate strong model predictions from limited D-Optimal screening studies where the purpose of these optimal designs is to maximizing the information for parameter estimates.
Comment: I understand how difficult it is to work within a constrained paradigm that makes the development approach more challenging than it needs to be. Developers should be allowed a bit more latitude when building their case for developing a new process with limited data and time.
Comments of risk assessment and management, subjective judgements, and other nonsense:
Tony Cox; A successful risk analysis estimates the causal relations between decisions and probable resulting exposures, and between exposures and their probable consequences.
Risk Analysis of Complex and Uncertain System, pp. 96.
See “Improving Risk Management Comparisons and Decisions,” a useful webinar by Tony Cox here:
My Comment: Qualitative risk assessment methods use ordinal values in place of implied probabilities because it is believed that these methods are more expedient and carry little loss of risk understanding.
Daniel Kahneman: Subjective confidence in a judgement is not a reasoned evaluation of the probability that this judgement is correct. Confidence is a feeling, which reflects the coherence of the information and the cognitive ease of processing it. It is wise to take admissions of uncertainty seriously, but declarations of high confidence mainly tell you that an individual has constructed a coherent story in his [or her] mind, NOT necessarily that the story is true. Thinking Fast and Slow, pp. 212.
A review of my assessment of your QbD software tool follows in the next post.
Like Reply privatelyDelete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Observations of QbD Software Tool:
– It was challenging for me to identify the risk-based questions from the operational questions for each stage of the process. For example, in the QTPP-CQA stage the primary question was: “How “relevant” is each CQA to each QTPP? I would not classify this as a risk-based question.
– I did not understand the purpose or the derivation of the percent values observed to the right of each CQA entry on the QTPP-CQA matrix.
– On the CQA-CPP matrix the primary question was: “How much does a Parameter (X) “affect” the Attribute (Y)? Again, this did not appear to me to be a classic risk-based question.
– On the CQA-CPP matrix there were percent values below each CPP which was not explained in the demo. I don’t know the purpose of these values or where they came from.
– On the “Occurrence” matrix the requisite question was: “How much will the process parameters go out of control, or be subjected to risk from …? The first part of this question asks for information that is unknown at the time of evaluation, because the process has not been developed yet! The second part of this question, which appears to carry some type of risk assessment guidance, is incomplete.
– It’s difficult to establish a value for the Occurrence column of the “Occurrence” matrix apriori…
– How is CPP Impact, CPP Relative Occurrence, and the ratio of these two quantities computed in the matrix? What is the purpose of these values?
– When viewing the Control Strategy tab it was not very clear to me the purpose of this chart. When we complete the “design space” DoEs we should have a good idea which process variables are significant control variables and which are not. As such, we have no need to use the chart on this tab to manage process understanding and control.
– Again, I could not find the use of risk-based questions on the Control Strategy to guide me accordingly towards a risk-based evaluation.
Parting Comment from W.E. Deming:
“Experience teaches nothing. In fact there is no experience to record without theory… Without theory there is no learning… And that is their downfall. People copy examples and then they wonder what is the trouble. They look at examples and without theory they learn nothing.” W. Edwards Deming in The Deming of America
I hope you find this posting useful. Keep at the software product Sun. I suspect it will evolve to become a very useful tool in the future.
Cheers,
Ken
Like (1) Reply privatelyDelete 1 month ago fereshteh barei likes this
Olivier MICHEL
Olivier
Olivier MICHEL
Senior Consultant chez ALTRAN AG
Ken, Sun et al, this is (was?) a tremendous discussion, thanks! would you mind me lighten a little bit the burden? Sorry it’s a bit french-touch, (translation on demand)
about knowledge, experimental design
– QbD to be jumped in at once: “Le premier était de ne recevoir jamais aucune chose pour vraie, que je ne la connusse évidemment être telle c’est-à-dire, d’éviter soigneusement la précipitation et la prévention”
– getting the right level of detail and structure: “Le second, de diviser chacune des difficultés que j’examinerais, en autant de pour celles qu’il se pourrait, et qu’il serait requis pour les mieux résoudre.”
– about getting the right knowledge and process understanding:” Le troisième, de conduire par ordre mes pensées, en commençant par les objets les plus simples et les plus aisés à connaître, pour monter peu à peu, comme par degrés, jusque à la connaissance des plus composés..”
– about missing CPPs: “Et le dernier, de faire partout des dénombrements si entiers, et des revues si générales, que je fusse assuré de ne rien omettre.”
In short: Good point Deming! Well done Descartes!
Unlike Reply privatelyDelete 1 month ago Ken Myers, Sun Kim like this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
I love answering this question because this software feature is the core of how we can correlate a process parameter to the patient mathematically.
There are 3 layers of data that scientists and regulators want to correlate:
QTPP (patient) – CQA (product specs) – CPP/CMA (controlled by operations)
In LeanQbD software, we do this step-by-step.
First, establish relationship between QTPP to CQA. (Ask: How much does this QA affect this TPP?)
Next, establish relationship between CQA to CPP. (Ask: How much does this PP affect this QA?)
Finally, we tie these together with simple linear algebra (multiplication and addition).
As you can see, this is very logical and mathematical.
Now contrast this with traditional FMEA where we try to achieve the correlation by asking, how likely is this process parameter to harm the patient? The issue with this question is that our brains can’t make such a far cognitive jump — pH in media vs duration time of lyophilization: which process parameter will affect the QTPP more? Now do this for 1000+ parameters…
In the traditional FMEA, we skipped a layers of data, hence trying to make a cognitive jump without the intermediate step. As a result, scientists feel fmea sessions are very subjective, tiring and only get mediocre results.
Notice how easier it is for scientists to answer:
How much does this QA affect this TPP? & How much does this Parameter affect this Quality Attribute?
These are scientific questions. We can express them as: Y=f(x) or QTPP=f(CQA) and CQA = f(CPP).
So as you can see, we just broke down the question into 2 logical steps.
The alternative is excel based FMEA which can NOT correlate the impact that a Process parameter has on the patient (QTPP).
Other industries have been using a similar approach. We just adapted it to our industry.
As for the calculation:
In the Modify Drug section, you assign a ranking/score to QTPP’s (default is medium)
In the QTPP-CQA tab, when you are assigning a ranking to CQA’s,
software multiplies individual QTPP ranking and individual CQA ranking. i.e. 3 x 3 = 9.
You do that for all QTPP and CQA combinations.
Now you have a total score of each CQA.
In CQA-CPP, you do the same for all combinations of CQA-CPP’s.
Now you have a total score of each CPP.
To compare apples to apples, the software normalizes (changes raw scores into relative percentages)
Normalisation is: individual CPP score / total score of CPP x 100% = Relative Importance of CPP
This goes same for CQA and Occurrence.
From: http://www.leanqbd.com/faq/
I hope this answer clarifies why “Correlation of QTPP-CQA-CPP” is important and how the software calculates it.
Delete 1 month ago
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Oliver, Merci pour les citations utiles! (excuse my broken French)
Delete 1 month ago
fereshteh barei
fereshteh
fereshteh barei
Strategy advisor, Business analyst,Health and pharmaceutical industry
Hi Sun, thak you for the message and alsi thatk you for launching this discussion. Many thanks to Ken Myers for his comments and the references that he provides they are always a source of inspirations and let me undrestand those technical subjects betters.
As for my article, I haven’t yet decided about the title it will come at the end, but it will be about reducing uncertainty around biosimilars and their value-added aspect, this is focused on innovation and how specific technical methods may help to decrease this uncertainty and why it may be promoted in the case of this treatments (as well as the others). I will tell you when it’s finished, I also look for the best journal to publish this one, any idea?
Best regards
Like Reply privatelyDelete 1 month ago
Ken Myers
Ken
Ken Myers
President/Principal at Ascendant Consulting Service, Inc.
Thanks Sun for your detailed answers above.
It appears you translate the textual inputs of Low, Medium, and High into ordinal values which are hidden in the meta-data of the software along with the calculations. The approach you use is similar to Quality Function Deployment for product development. Let me see if I can follow the correlation between your QbD development approach and the approach used by other industries supporting Quality Function Deployment (QfD):
QTPP assessment in QbD (is equivalent to) Product Planning (Phase I) in QfD,
with inputs being customer needs, the process being to establish key product performance measures, and the outputs are target values for the product performance(QfD and QbD) for parts (QfD) or quality attributes (QbD) — which at present I don’t see in your tool. The drug development scientists should be responsible for this stage of the development process, and inform the manufacturing team of their developments to insure their design direction is moving towards the development of a manufacturable process.
QTPP to CQA assessment in QbD (is equivalent to) Design Deployment (Phase II) in QfD,
With inputs being the critical key product measures (QfD) or product quality attributes (QbD) and target values for each part (QfD) or each quality attribute (QbD) from the Product Planning stage, and the process is to establish design criteria for the product design, and outputs are to identify the critical part characteristics (QfD) or Critical Quality Attributes (QbD). This is a shared responsibility between the drug development scientists, manufacturing engineers, and quality assurance team. The scientists should take the lead role in this phase of development, but recognize the manufacturing team is their customer. The goal is to develop a drug product that is manufacturable…
CQA to CPP assessment in QbD (is equivalent to) Process Planning (Phase III) in QfD,
with inputs being the Critical Part Characteristics (QfD) or CQAs (QbD) and manufacturing process constraints, the process being to identify the Critical Process Characteristics for manufacturing parts (QfD) or for manufacturing the drug (QbD), and outputs are the identification of critical process characteristics (QfD) or CPPs (QbD) and their target values along with operating windows and requisite control plans to maintain high process performance. This is a shared responsibility between the scientists, manufacturing and quality teams with the manufacturing team taking the lead role at this phase of development. The manufacturing team should recognize that the quality team is their customer. The goal is to develop a quality manufacturing process that meets all of the established CQA requirements, thereby meeting all of the QTPPs.
Production Planning (Phase IV) in QfD and QbD, with inputs from all preceding phases of product and process development, the process being to develop all of the operating controls in the business needed to manufacture the drug product, and the outputs are operations instructions, equipment maintenance schedules, material logistics plan, quality control plans, lean ops plans, etc. This is a shared responsibility between the manufacturing, quality, production planning, and maintenance teams. Both the manufacturing and quality teams should take a shared lead role in this activity. They should recognize their customers are all of the business units that support the manufacturing operation.
Hopefully I got this flow right per your QbD tool. I still have some concerns on its operation which I think is has too many degrees of freedom, and possible limitations on the risk management approaches. But, I’m beginning to understand it.
Thanks for your time in answering my questions about your QbD tool.
Like Reply privatelyDelete 1 month ago
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Ken, I’m glad it was helpful. Inspired by QFD and modified it to specifically fit QbD needs. Yes, this approach encourages collaboration among development, quality, manufacturing and operation.
Delete 1 month ago
Alicia Tébar
Alicia
Alicia Tébar
Manager QA & QbD en Azbil Telstar
Yes, excellent discussion. Thanks to the rainy Sunday that gives me the time to read everything.
When I first check Sun’s software I missed uncertainty. The reason I gave to him is that in development I really don’t know how to apply occurrence without data. The results in my opinion have a lot of subjectivity.
I prefer to apply uncertainty like a kind of gap analysis in order to visualize in my process what do I know and what do I don’t know.
This is just a necessary starting point that don’t take you many time, then go to DoE, go to the lab/pilot plant and let’s have “real” data.
In summary, and in my opinion of course, the most efficient approach is: mapping the process in detail, identify “candidates” to CMAs and CPPs (CQAs already defined), introduce those variables in a risk table with clear criticality and uncertainty scores and then go to DoE. With the results then going back to risk analysis and go on.
I don’t like to dedicate much time to complex risk tables, at least in development stages.
A different situation is when you need to approach the PPQ, is then when you need and when you have enough information to prepare a really good risk analysis table (probably FMEA).
Unlike Reply privatelyDelete 1 month ago Sun Kim likes this
Sun Kim
Sun Kim
Sr. Manager, Master Black Belt in Quality-by-Design, Agile Lean Six Sigma and Design Thinking
Top Contributor
Alicia, thanks for requesting this feature for Lean QbD. We are trying our best to continually improve the software based on real feedback like this.
Delete 1 month ago
Sidney BURFORD
Sidney
Sidney BURFORD
Freelance Consultant at Regulatory Affairs CSO
Impressed with this excellent and informative discussion, thanks particularly to the contributions by Sun and Ken.
My feeling is that, for many small molecule NCEs, most companies will continue to have a fairly informal risk evaluation process for the early stages of process development – but the people involved have the important responsibility of identifying all the variables (always difficult to recognize one’s assumptions!) and determining their importance and the levels of uncertainty/unpredictability.
For later stage pharmaceut
It’s an awesome post for all the web visitors; they will get benefit from it
I am sure.