FDA Reviewer Reveals Tips on QbR for Drug Substance
Are you in the Generics industry? Then you should be familiar with QbR for Drug Substance. Barbara Scott of OGD (Office of Generic Drugs) at FDA shares insider’s tips on how to successfully file a Question based Review submission.
She breaks down the questions regarding Drug Substance in Sections
- S.2.2 Description of the Manufacturing Process and Controls
- S2.3 Control of Materials
- S2.4 Control of Critical Steps and Intermediate
- S.2.6 Process Development
and then kindly points out the expected answers for the questions from the FDA reviewer’s point-of-view.
She talks about:
- Must-have information in your Question based Review
- Examples or templates or tables for the specification on starting materials.
- Common mistakes reviewers find in the submissions
I bolded the tips in her talk.
—- Here are my notes (*may contain typos)—
Barbara has been with the FDA since 2004, and she’s currently the Acting Team Leader in OGG. Barbara has played a huge role -in drafting the questions for the drug substance.

Barbara: I’m going to be focusing on the question-based review quality overall summary for the drug substance. And some of you may have attended the GPHA meeting, or recognize these slides. They were presented by Carolyn, Deborah and Ram at GPHA. For those of you who weren’t there, they’ll be brand new… (laughing). And…I’ll just issue the standard disclaimer. These are my thoughts and opinions, and don’t necessarily represent FDA policy.

The general outline for my talk today is, we’ll first talk about some general expectations of the QbR QOS for the drug substance. And then, I chose Section 2.2, 2.3, 2.4 and 2.6…out of the CTD for the drug substance to highlight what the questions are, and what our expectations are for your response. So, as…Dr. Sumitha said, the question-based review QOS has design with expectation that the drug substance application, whether or not it’s coming in as a…as a drug master file…in a NDA submission, or if the ANDA holder is submitting the drug substance information. The expectation is that…a quality overall summary will be submitted, okay, and this will be in module two and filed along with CTD format.

They’ll be twenty-four questions, and the question boxed, followed by the firm’s response. And it covers sections S-1 to S-7. And this is an important thing. The information in Module Two…needs to be consistent with what’s found in Module Three. The holder applicant is encouraged to refer to the draft and final agency guidance. Especially ICHQ-11 when it comes to the drug substance. And, if a question isn’t relevant to your drug substance manufacturing process, for some reason…please don’t just leave it blank. We don’t know if you didn’t see the question…or what. So, just say, “Not applicable,” and follow it up with a brief explanation as to why.
S2.2 Description of the Manufacturing Process and Controls

Moving on to S-2.2, this is the description of the manufacturing process and controls. There are…two questions under this section, and the first one is, What is the flow diagram of the manufacturing process that shows all incoming materials, reagents, reaction conditions and in-process controls, and, if appropriate, any re-processing, re-working or alternative procedures? So, our expectation for a response with this would be a complete synthetic scheme, with chemical structures, reaction conditions and reagents. All of you have read Jack’s papers; you’ve all read Joe’s papers, so we want to see what you would submit for a Jack’s paper. Okay, not vague, just an arrow going from reagent A to B with…with no conditions, okay? We want a brief summary of the manufacturing process that includes the batch size, input materials, molar equivalence…and expected yields, your actual yields…and percentage yields for each step. Isolated intermediates should be clearly designated, and the converse to that is if you’re not isolating intermediate, please put it in brackets, okay? Brief description of the re-processing and re-working procedures if you have them, and when to apply those procedures. And you can…reference supporting data for that.

And then this question came specifically directed to this audience for…PAT. So, applicable, where on-line, at-line or in-line technologies are proposed for routine commercial production that allow for real-time process monitoring and control. And provide a summary of how each technology was developed. So, to be honest we don’t see all that much of this in drug substance manufacturing, not yet, so…hopefully that’s going to change. But, what we would like to see here, and when you do make your submission and answer this question, is a description of the online, at-line and in-line technology, and a traditional method that’s being replaced. And then part of your risk assessment (ban) for this would be if it’s a medium impact, we want to know…you know, just a short summary. But if it’s high impact, we want to know…have a description of the instrument, it’s location, the development, all the validation. So, you have to be a little bit more…forthcoming with…with all the information that went into the development of using that technology. And, you’re all familiar with PAT.
S2.3 Control of Materials

Moving on to 2.3, control materials. There is…three questions in this section. And the first one is, what are the starting materials for the manufacturing process and how would changes in the starting material quality or synthesis/source be controlled to minimize adverse effects on the drug substance quality? By far, this is probably the biggest sticking point I see in my DMF reviews, okay. What’s the starting materials? Proposed starting materials should be clearly identified with appropriate specifications. And the justification for the starting material should be provided, and I’ll refer you to ICH Q11. I’m going to talk a little bit more about this when it comes to the…process and product development report. But, in the minimum, in the QOS it needs to include the name address and…contact information of the manufacturer and a flow diagram and description outlining the synthetic route and conditions of each proposed starting materials. And this is really only applicable if you’re…designating a late stage intermediate as your regulatory starting material.

So, keeping that in mind, the next slide discusses what else we want to see when those late stage intermediates are what you’re proposing as a regulatory starting material. We want to see a discussion of the impurities, including the residual solvents and inorganic impurities, arising from the manufacturing process of each proposed starting material. We want to know that your analytical procedures are capable of detecting those impurities, and we want to know the fate and purge and…of them and their derivatives in subsequent downstream processing. How does the proposed specification for each starting material contributes to the overall control strategy? You can reference that, you know, to your process development report, 2 6.
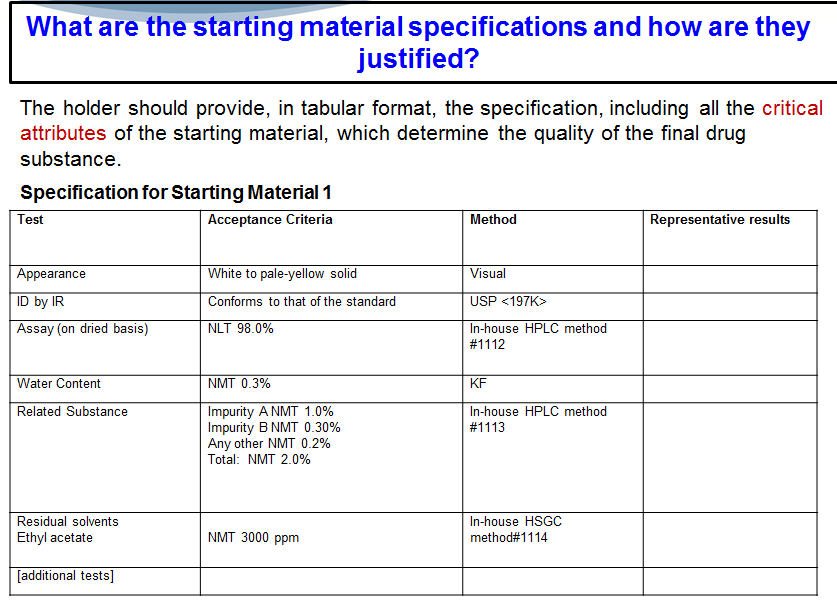
The second question is, what are the starting material specifications and how are they justified? Again, this is pretty standard stuff, right? We’d like you to provide a table for each starting material that includes a test, the acceptance criteria, the method you used and the representative results.
The next question is, what are the specifications for reagents, solvents, catalysts, etc.? And, what are the critical attributes for these materials that impact the quality of the final drug substance? So, when you’re considering your in-action scheme you want to make sure you tell us all the materials, what’s the process stage they’re…used in, and you can just give a simple reference here in this table format. Remember, this is your quality overall summary, okay? So, you’re directing us to where to look, so we don’t have to hunt and peck…for where this information can be found in Module Three. And then, of course, if plant or animal-based materials are used, where the BSE, CSE certification can be found. And then along with that, you need to provide us the critical material attributes that you have identified as important to the quality of the final drug substance. And link the process steps and the CMS together.
So, if you just consider a grain yard reaction, we all know water is important to control. So the solvent and the magnesium would need to have the water control. So, just…provide us a table and say what the stage was, the reagent or solvent, the critical material attributes, which is water in this case, and its impact, okay? Again, just a nice summary table, that’s all you need.
S.2.4 Control of Critical Steps and Intermediate
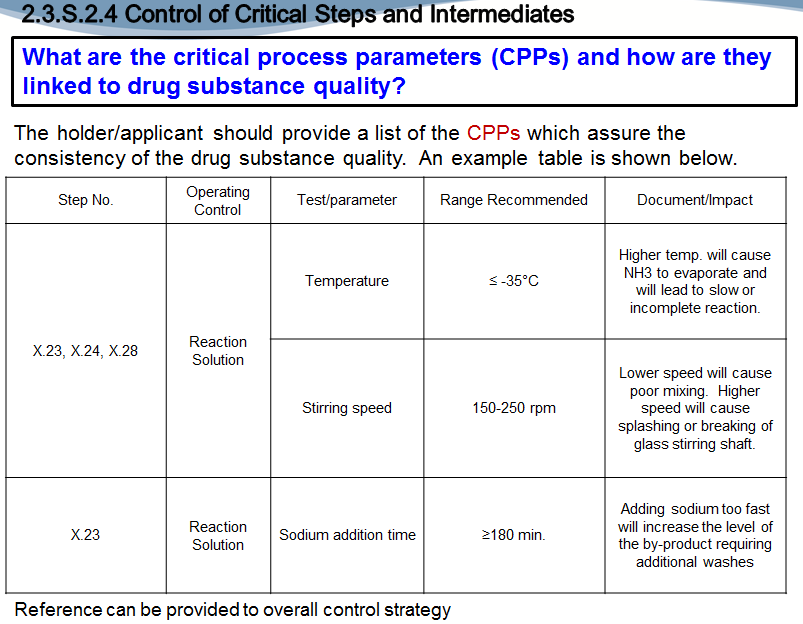
And…2.4, Control of Critical Steps and Intermediates, there are also three questions. What are the critical process parameters and how are they linked to drug substance quality? So, our expectation again here and the quality overall summary is a nice table. It lists the step number…the operating control, the test parameter, the recommended range and the impact. So, in this table you can see that…step X.23 has three critical process parameters, three recommended ranges…or a range recommended for each, and then a documentation as to why that’s critical to the overall quality of the finished product.
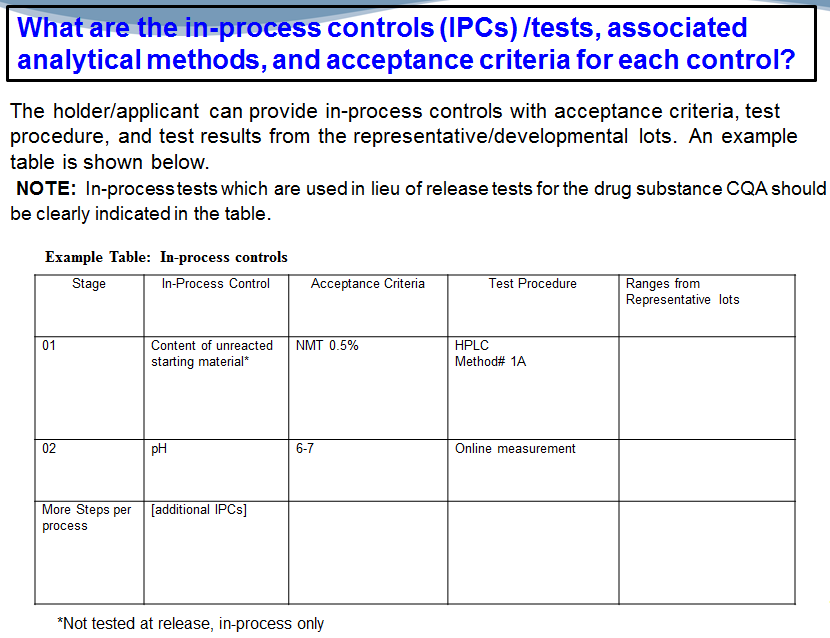
What are the in-process controls and tests associated with…associated analytical methods and acceptance criteria for the in-process. So, again, table…(laughing)…showing the stage, the in-process control, the acceptance criteria, the test that you used and ranges from representative lots that you’re providing in your executed batch record, right? You should also note if you’re…you’re testing at release or not testing at release, and you’re only testing something in-process, you can make a note of that here as well.
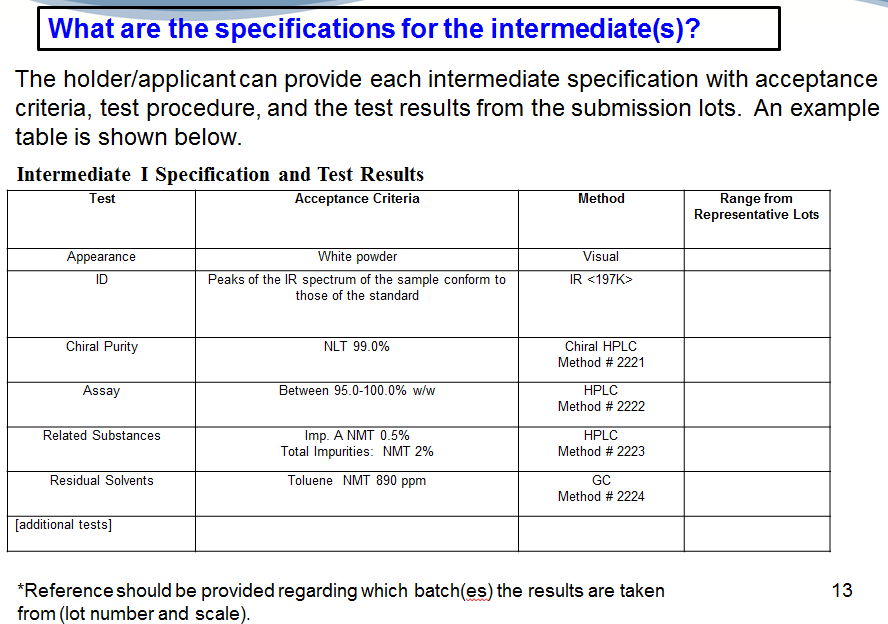
And then, what are the specifications for the isolated intermediates? You can provide…for each intermediate, a table with the acceptance criteria, test and test results. Does this sound familiar?…. (laughing)… Sorry to be so…repetitive, but I think sometimes when you can see it clearly outlined…it’s much easier for you to put it together for us later. And, definitely when you…provide us this table get a little footnote at the table of where these results are coming from, what the scale is of the batch, okay?
S.2.6. Process Development

And then lastly, and maybe deceptively…so, there’s only one question under 2.6…Process Development and that’s, what development and scale up information supports the commercial process and control strategy? So, we’ve been talking a lot about risk, a lot about control strategies and…and for the drug products, and the talks I’ve been going to, and the same thing applies to the drug substance, okay? Really, it’s the same principles. And the goal is that, for the drug substance you want to consistently obtain an API of intended quality. And so, what we want to see in 2.6 is a summary overview, maybe twenty pages and it should include your scientific understanding of your process and can be based on literature, the patent literature, your in-house expertise, it should discuss the drug substance critical quality attributes. It should explain how is the process chosen, how did you get from A to B, why did you choose A to start. Your approach to development, did you use traditional development or maybe some enhanced technology and a description of the control strategy.

The relevance of the development studies to scale up, what scale have you done this on and can you do it reproducibly, and then, tie the knowledge gained back to the drug substance Critical Quality Attribute. And where does the Critical Quality Attributes of the drug substance come from, prior knowledge and that communication that gets fostered between an API supplier and your client. Okay, so I review mostly drug master files for the…for the (underside) of…of drugs here. So we’re used to seeing…multiple synthetic routes for the same drug substance, multiple…you know, one DMF holder has multiple clients. So as the DMF holder, they’re responsible to know what their client needs, and so, there’s got to be communication between the DMF holder, the API supplier and the generic or the Pharma industry on what they require. So open that communication line. What are the characteristics of the API that are required? Is it sterile? What’s the impurity profile that’s acceptable? Is polymorphism an issue? Is particle size an issue? And notice the Critical Quality Attributes aren’t necessarily the same thing as the specification. So we want to know, bottom line, is how did you determine the CQAs and what’s their justification?

So, as I alluded to, I was going to talk a little bit more about starting materials and where does the manufacturing process begin. Well, from the beginning right? And that’s with the designation of the Regulatory Starting Material. I’m just going to put these references up. ICH Q11, outlines our current thinking on what an appropriate Regulatory Starting Material should be, and these are the pages. ICH Q7, you know, just reference to GMP right? There’s a nice table in Q7, that associates risk with the number of steps before the finished products. So, we refer you to that table. The further back you are the less risk. And then Q10, again that same thing is for the drug…drug product, to be…Critical Value Attributes, okay. So the selection of the Regulatory Starting Material…should consider the following general principles, okay, and you need to think about them all in concert. And again, this is my allusion to GMP, changes in the early steps, upstream process have much slower impact on the quality of final API. So, think about the number of steps you might need to have, before the final API to make sure you have a good control. Describe enough for us to understand how and where the impurities in the API are formed and why the proposed Control Strategy will ensure that. The steps impacting the impurity profile should normally be included, and so this goes back to changes in the early steps, okay. If you’re genotoxic impurities are far upstream, they’re probably okay, but if they’re in the middle or right before your Regulatory Starting material, then we’re probably going to have a problem with that. Okay, we want those under GMP.
If you have a convergent synthesis, make sure you…it doesn’t meet both starting materials and this core kind of go with number six. I think there’s a lot of confusion in the industry and what we mean by significant structural fragment in this context. We…we’ve had people submit drug substance DMR files and you have the final intermediate and they say, “This is okay, because there’s a significant structural fragment. It is the…the structural fragment.” So, what we mean is if you have two halves of a molecule and both of those halves are starting materials, okay? So, if it…if whatever you’re starting in a convergent synthesis, if A contributes a significant structural portion of overall molecule that is the starting material and it needs to be declared. And then obviously, in situ intermediates, that aren’t isolated cannot be a Regulatory Starting Material because there is no isolation.

So once you have your, starting material designated from a regulatory perspective, and the synthetic route outlined, you move on to the Manufacturing Process Development. The optimization studies what changes you might…you want to describe significant changes from lab to production scale.

Discuss the approaches used to optimize the process and identify the Critical Process Parameters within that (route) synthesis and what the control strategy is that you have in place. And you can use different approaches to process optimization, okay, this is what we’re talking about here. You can use a traditional approach, where you have a defined set of points and operating ranges…where the joint substance control strategy is basically based on the demonstration of process reproducibility on the scale that you’re proposing. Or, you can use an enhanced approach, using PAT, okay, and then again, I’m just… we haven’t seen a lot within…within our industry on the drug substance side. But you can select…an enhanced approach certainly…where the risk management and more extensive scientific knowledge is required in order to…select appropriate process parameters and unit operations and then, of course, corresponding studies to support that. And you can use either or both…it’s up to you. But you need to tell us what you’re…what you’re doing.

Sure you’re all aware of this definition. You’ve seen it probably…I’ve seen it at least three times since being here the last couple of days. But, you know, that control strategy that you hear about for drug products also applies to drug substances, so just bear that in mind. And I like this one and that the control strategy is a comprehensive plan for ensuring the final product meets critical requirements, and therefore the needs of the patient. It’s always the needs of the patient that you need to be thinking about.
Further, the control strategy assures for the drug substance, that the process performance and product quality or risk management is low…or well, process performance is high and risk management is under control. And it’s…and it’s important that you have a control strategy for every process. Whether or not it’s traditional of enhanced, or some combination thereof. And it needs to include all the material attributes, starting materials, raw materials…important process parameters, order of addition, the molar ratios that might be required to drive a reaction forward…purification steps, and what the in-process controls are. And those all relate then to what the specifications finally (………).
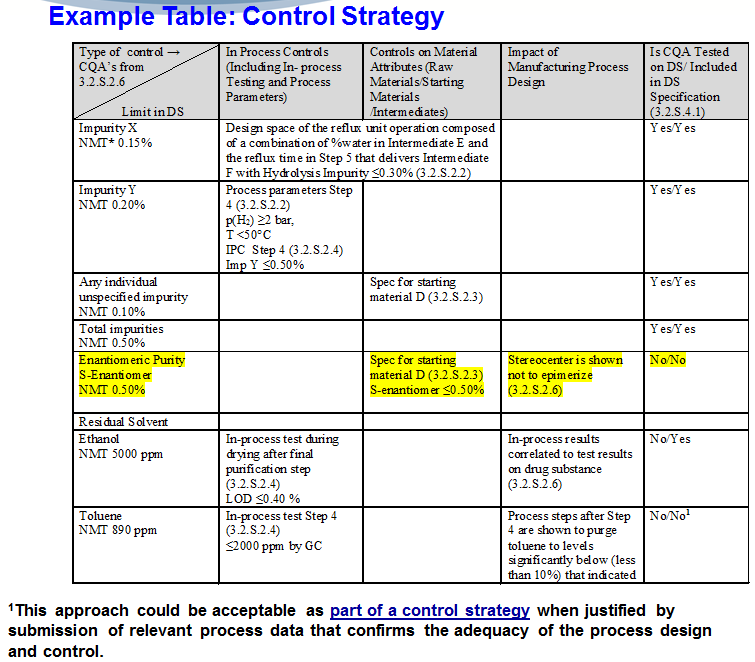
So, this table comes right out of Q11. So, you can take a picture of it, but… (laughing)…it’s pretty much right out of Q11. So…and it’s just an example of…of a control strategy. So, it has the type of control, the critical quality attribute, the limit in the drug substance, the in-process controls, the material attributes, the impact of the manufacturing process to design, and then, is the critical quality attribute tested on the drug substance or included in the drug substance specification. So, let’s just take an example where you have a (……..) APF, okay? You would expect an (……………….) to be a critical quality attribute, and indeed…that (……………..) in this case is being controlled in the drug substance at not more than point five percent.
So, this firm control strategy, they decided to put a specification for starting material…right in the…right in the first step, in controlling the S-Enantiomer right at not more than…0.50%. And then, in their product development they show the stereocentre doesn’t (…………….). So, therefore, in their control strategy, is a testing on the drug substance? No. Is it included in the drug substance specification? No. It is tested upstream.

So, here we get a clear rationale of why the firm is doing what they’re doing. And then, as a reminder you’re required by law to validate your process for APIs…prior to commercialization of the …of your client…from your client. Just a reminder. And this provides assurance of drug substance quality.

And then, finally some notes and links here. All the QbR/QOS drug substance questions that I skipped, that are in the CTD format, okay, can be found at this link. And we strongly encourage electronic submissions wherever possible. For the DMF staff, I can honestly say I…my upper body strength is really… (laughing)…increased, because most of our DMF solutions are still paper. So, please submit electronically if possible. And then if you have any specific (……) DMF questions for…for generics…or just in general, please feel free to shoot us an email at dmfogd@fda.hhs.gov, and someone will get back to you, okay? So, that communication between us and you is…is really important, and we do encourage you to ask questions up front. It might save us all some time, right? And finally, I’d just like to acknowledge…Dave Skanchy, he’s the…DMF…Director…Acting Director…Susan (Rosencrance) and Lawrence Yu for their leadership…the entire DMF Staff, we’ve been hiring like crazy, and are almost all the way up to staff at this point. And then finally, just…just thank all of you for your attention and for sticking around.
Other References:
CMC Initiative Development for New and Generic Drugs (Yu, FDA)
—————————————————————————————-
Thanks for reading.
Here are 2 other articles related to Question-based Review
FDA Reviewer’s Checklist: QbR for Drug Products