FDA Reviewer’s Checklist: QbR for Drug Products

What does a FDA reviewer look for in a (Question-based Review) QbR for Drug Products? Jennifer Maguire PhD from the Office of Generic Drugs (OGD) reveals her checklist for ANDA. In this post, Dr. Maguire shares:
-
What information all applications should contain.
-
What are the key questions that FDA reviewers will ask during the review.
-
How QbR questions map to Drug Product attributes.
-
How to determine a CQA and connect it to clinical performance.
-
How you should select the parameters and why they are important
-
Examples of Control Strategy
-
What to include in your DOE (Design of Experiments) studies.
So here are my personal notes:
Session Chair: This is Jennifer Maguire from the Office of Generic Drugs. Jennifer has been with the…FDA since 2010. She is with the OGD. Among her many roles in OGD, she is also the Quality by Design Liaison with the OGD. Jennifer has been one of the very active contributors in developing the QbR for the drug products. Jennifer…

Jennifer: So, I like to start with this quote…on what is quality. It’s from the book, “I Know It When I See It.” I’ve never read the book, I just love the quote. So, our customers tell us when we have a quality problem, we turn to our specs and our tolerances to see if they’re right. Customers aren’t interested in our specs. They’re interested in the answer to one simple question, “Do the product do what I expected it to do?” If the answer is yes, then it’s a quality product. If the answer is no, then it isn’t. At that point out specs and tolerances aren’t wrong, they’re just irrelevant.

So, the reason that I like this is one of the things that we’re really doing with the revision of QbR, is making sure that we have a very patience, centric, focus, I think this quote really speaks to that.

So…at the heart of QbR are, sort of, fundamental questions that we as regulators are thinking about. So, will the product design and show the desired performance? Will the applicant be able to scale up to commercial scale and ensure comparable quality to the bio batches? And will the applicant be able to manufacture the product, which defines quality parameters over time? Okay, so hopefully most of you are familiar with this (cedar-map), this is manual policy and procedures.

This particular map deals with…how the principles of ICH Q8…Q9 and Q10 should be applied to CMC review. So, it’s written for reviewers, but it does say that all application should contain the following: so, quality target quality profile, the identification of the CQAs, selection of an appropriate manufacturing process, information that can (be as) understanding of the development of the drug products and its manufacturing process, identification of those aspects that are critical to product quality, and justification for the control strategy. And so, all of these things really drive the QbR questions.

So, the QbR questions for drugs products follow CDT. Those questions in each of these sections, and I’m not going to go over them all…in the essence of time, I want to keep you all awake. So, there is approximately thirty-eight questions right now. You can see here, I’ve…told you how many questions are in each section, and the bulk of them are in pharmaceutical development.

So, I’m going to show you couple pharmaceutical predominant questions today, and then…one or two from manufacture and control of drug product.

So, starting with pharmaceutical development. First question is, for a 505b(1) application, what’s the rationale for selecting the proposed dosage form for the drug product? For a 505b(2) and 505(j) applications, what are the characteristics of the listed or the reference listed drug product? What’s the Quality Target Product Profile of the finished product based on the proposed indication and patient population? How is the QTPP justified? So, it’s short and sweet.

The first thing we’re looking for a 505b(1) is the scientific and clinical rationale for the selected dosage form. For 505b(2) and 505(j) application, we’re looking for clinical, pharmacokinetic drug release properties and physicochemical characterization of the RLD product. A lot of this information can be pulled from the RLD label. As far as, you know, the composition of the RLD, we like your best guess. So you can usually achieve this through the labelling, reverse-engineering and patent literature gives you your starting point.
QTPP considerations include, but of course, not limited to…intended use and clinical setting, patient population, dosage form and strength, route administration, delivery system, container closure system release or delivery of therapeutic moiety, and…attributes that affect pharmacokinetic characteristics, quality attributes and alternative methods of administration.
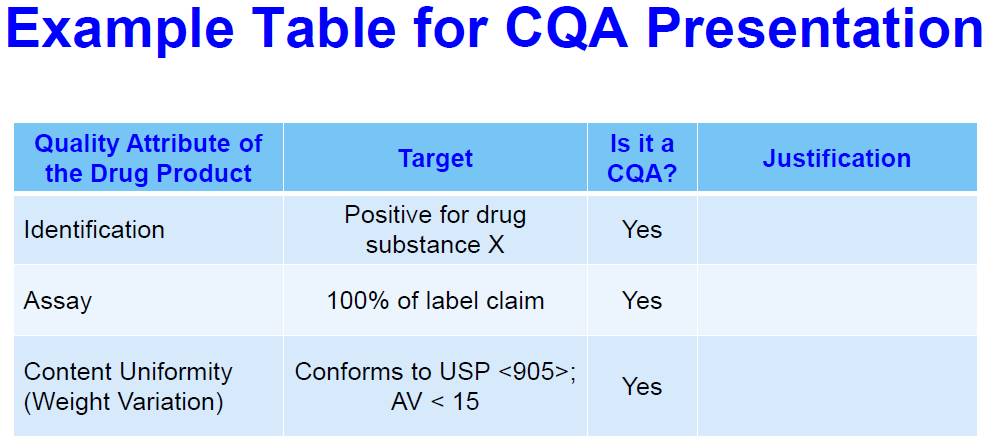
The next question, what are the quality attributes of the finish product? Which quality attributes are considered critical quality attributes? For each CQA, what’s the target and how is it justified? So, here we’re looking for a complete list of the quality attributes that are relevant to the dosage form of the drug product. In that list each of the attributes should have a target that should be preferably quantitative, and should be supported by development data and scientifically justified. These targets should be defined early in development based on drug substance properties, dosage instructions and intended patient population and that characterization that you’ve done for the RLD when that’s applicable. It’s okay if your target changes as development goes on, but then you need to provide justification for that. The risk based justification to identify CQAs should be based on the severity of harm to the patient with respect to clinical safety and efficacy and not on the probability of occurrence. So, here is an example table for…presentation of CQAs. So, in the column you have all of the quality attributes followed by their target. A simple yes or no is it a CQA and what’s the justification you use to make that decision.

The next question, what is the approach for meeting the CQAs related to clinical performance? If applicable, what in vitro bioperformance evaluations, such as dissolution method, flux assay, etcetera, (leads) to pharmaceutical development to ensure clinical performance. So, if you were looking for discussions of CQAs second impact clinical performance. The clinical effect can be direct or indirect depending on the pharmacological and toxicological determinants of the desired therapeutic performance. So, this list is not exhaustive, but just to give you an idea, therapeutic index, time to onset and time to loss of clinical effect, duration of therapy whether it’s acute or chronic, the need for titration, if it’s pro-drugs, if they’re metabolites…immunogenicity, so on.

So, some quality attributes that may impact the list of determinants that I just said, would include strength or potency, purity, as well as impurities, content uniformity, sterility or bioburden, endotoxins or pyrogens. The relevance of the dissolution test as a predictor of in vivo performance should also be discussed. Predictive dissolution methods are strongly encouraged for all BCS class two drugs, as well as all modified release formulations. If you’ve done any pilot bioavailability or bioequivalence studies we ask that you give us a description of this, as well as anything that you have learned from those studies and how it’s changed your formulation or your dissolution method development. A dissolution method development history is very helpful for us in understanding the…drugs product formulation decisions that you’re making and the discussion of any other in vitro methodologies and they’re relevance as predictors of in vivo performance for the more complex dosage forms.
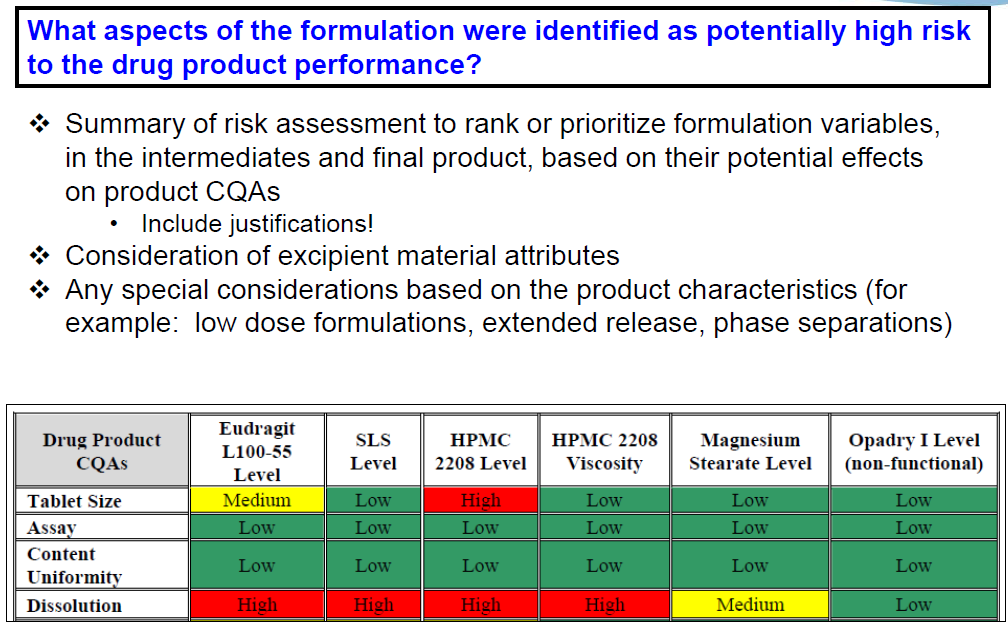
So, the next question. This is a formulation development question. What aspects of the formulation were identified as potentially high risk to drug product performance? So, here we don’t dictate what type of risk assessment tool you need to use and we don’t ask that you do informal versus formal risk assessment. Those are decisions that you make. But what we are looking for is a very concise summary of your risk assessment outcome. It helps you rank or prioritize your formulation variables in both intermediates and final product, based on their potential effects on the drug product CQAs. So, I’m showing you here an example table…that we’re actually seeing a lot of now in generic applications. Where you use three colors to represent low, medium and high, and then this table will be followed by another table or text that clearly explains what the justifications behind these decisions are. Especially since anything that’s low presumably you’re not going to do further studies, so we want to know your rationale for those decisions.
We ask that you consider excipient material attributes. Not just levels of excipients, and really, you should consider anything…any aspect of the formulation that’s going to impact the drug product performance. So, some of those special considerations maybe of low drug loading…the level of polymer for extended release formulation, phase separations, things like that should all be assessed.

So, this next question is a…process development question. So, before this question in the QbR, there a question that asks for the rationale for your manufacturing process selection. And there is the question after that that asks for the overall risk assessment of your manufacturing process that you’ve selected to identify steps that are more the high risk steps, and then it goes to this question. So, for each of the potentially high risk manufacturing unit operations, and then there is four parts. Part A, what input material attributes and process parameters were selected for study? And what are the justifications for their selection? So, if we were asking you to identify intermediate CQAs that impact the drug product CQAs. And then once you identify this intermediate CQAs, we ask you identify all the material attributes and process parameters that can then impact those intermediate CQAs. Use risk assessment to narrow down the long list into the vital few. This really focuses your development efforts, and then in cases where you’re going to reference prior knowledge, that’s fine. But we need to know the source of the prior knowledge and we need you to establish why that knowledge is relevant to the current process. So this should be very clearly described.

Part B of this question, what process development studies were conducted? Provided a summary table listing batch size, process parameter ranges, equipment type and estimated use of capacity. So, here we’re looking for a summary in some sort of manner, that would allow the reviewer to follow the logical progression of your drug product development. So, for each study that you do it would be nice if we got the objective of the study, the design of the study, and the scale that it was performed at, factors that were investigated, responses that you measured and how that data compared to the pre-established targets you were shooting for, and the overall outcome of the study.

In the case where you do DOEs…we look that the conclusion is presented in a way that we see that the study was statistically meaningful. It’s helpful if you provide your alpha value for ANOVA…ANOVA tables. You must show that the model is significant, and that the lack of fit is not. If you’re using the center points then we will expect some sort of discussion about the curvature, if there is curvature…or if there’s not. And then, visual aids are really helpful. So, if you’re talking about significant factors, half normal plots…(……) charts are helpful, main effect plots, interactions, contours plots that show how two factors (jointly) affect your response, overlay plots that show how you’ve come to determine what your proven acceptable range is. They say a picture say a thousand words, and in this case it really does. So, we encourage you to use those graphics. If you choose not to do DOE, then we will ask that you clarify what significant means, and we would ask you to discuss how decisions regarding criticality are made. And then for complex drug products you should really consider verifications studies performed on the larger scale.

Part C, what process parameters and material attributes were identified as critical? And how do they impact the drug product CQAs. So, this is pretty simple to answer. But your answer really conveys your process understanding. So, it’s a very important question. The reviewer will ensure that the proposed commercial control strategy is supported by the development work. So, again, your answer to this question very important.
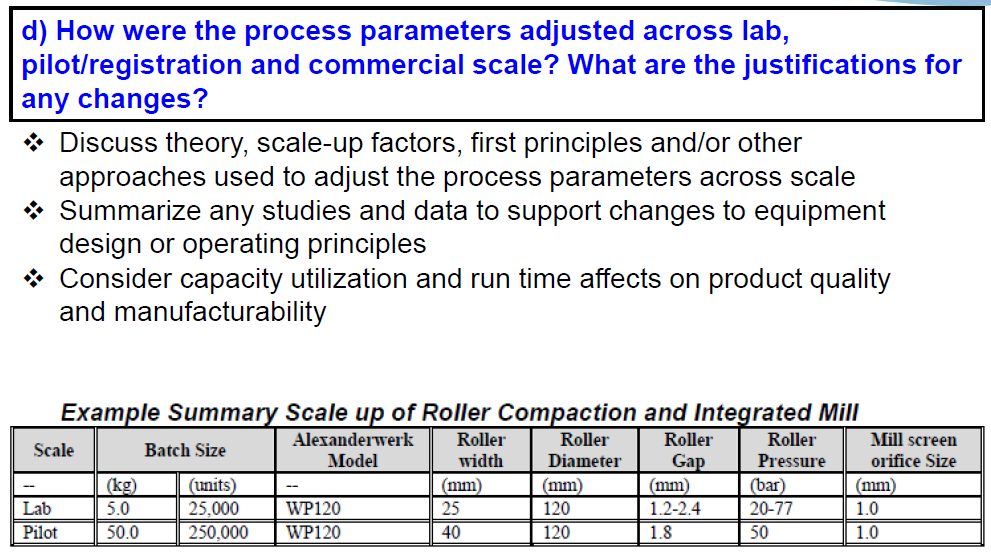
And then part D, how are the process parameters adjusted across lab, pilot or registration and commercial scale? What are the justifications for any changes? So, here we look that you’ve discussed and theories, scale-up factors, first principles, and any other approaches that you use to adjust your process parameters across…scale. Should you decide to change equipment…to…equipment with the different design or operating principle, we’ll be looking for a summary of any studies that were conducted to support this change. And then, we ask that you consider capacity utilization and run time affects on product quality and manufacturability. Then I have just a table here showing…an example of how you might present your scale up of the roller compaction and integrated milling, which shows how the settings change from lab to pilots scale.

So the next question I actually won’t spend too long on. We ask this question both for drug substance and drug product. But just to reiterate what Barbara said, obviously when you’re…online, atline or inline monitoring technology is more of a high impact technology, then the…the amount of information we would ask you to submit increases substantially.


So, moving to P.3 Manufacture, what is the detailed process description including process parameters, material attributes of raw materials and intermediates, equipment type, batch size, in-process controls including acceptance criteria and any proposed reprocessing? So, here we ask that you submit either proposed master production batch record, which we receive in most (……………) submissions or in the NDA world…narrative that is…at comparably level of detail that includes…and again, this is not an exhaustive list, but, batch size, incoming raw material or intermediates and acceptance criteria, the raw material grades, your in process controls and acceptance criteria, equipment type, vendor, etcetera.

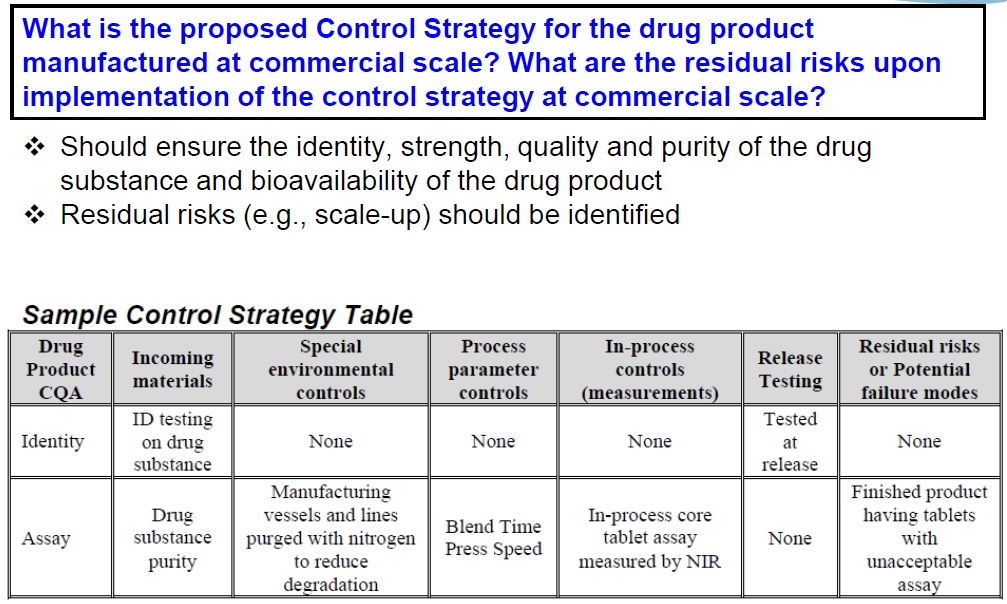
So, P.5 Control of Drug Product, we ask what’s the proposed Control Strategy for drug product manufactured at commercial scale? What are the residual risks upon implementation of the control strategy at commercial scale? So, your control strategy should ensure the identity, strength and quality and purity of the drug substance and bioavailability of the drug product. And we’re looking here that you…be forthcoming with any residual risks, for example, any scale-up risks…to really even our review. So, here I have a simple control strategy table, where on the left column you have the drug product CQAs. So then, let’s just look at one. For assay, for incoming material…the drug substance purity obviously will affect that assay. Special environmental controls, we have manufacturing vessels and lines which are purged with nitrogen to reduce degradation. Process parameter controls that would be relevant, so blend time, press speed…I’m not even…kind of read the…the rest of that. But you get a flavor for what we’re really looking for in here.

So, just to summarize. Question-based review is an established quality assessment tool that focuses on critical pharmaceutical quality attributes. I say it’s established because it’s really improve the quality of generic application, and we’ve been doing this since 2007. It’s transformative, I think it has the potential to…really make both brand and generic applications…modern and risk-science based approach. It’s a pathway for demonstrating product and process understanding. And it’s a way of increasing transparency between industry and the agency.

I hope this helps on your preparation for the next QbR for Drug Products. My question is how will a QbR look like for Biotech / Biologics products?
2014.Nov. FDA updates QbR to reflect QbD principles.
http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ManualofPoliciesProcedures/UCM423752.pdf