QbD – Doing More with Less with these 3 Tools [Mehtap Saydam]
What if we could predict the relationship of QTPP, CQA and CPP of our drug – with less clinical data?
Mehtap Saydam, our QbD evangelist from Turkey describes that there is a way with the 3 tools –
In Vitro Testing; Biopharmaceutical Classification Systems; and QbD Risk Assessment.
“In vitro dissolution testing, together with BCS (Biopharmaceutical Classification Systems) considerations, could provide a key link between manufacturing/product design variables and clinical safety/efficacy in QbD. Current dissolution methods are based on providing an acceptable risk level as proven by long-term experience rather than modeling the in vivo situation. More in vivo–relevant in vitro test methods would allow more flexible approaches than described above, further reducing the need for additional in vivo studies.
In addition, the use of algorithms that model the absorption process in a more mechanistic manner than simple IVIVC approaches would aid the establishment of drug specific definitions of “design space” and to minimize bioavailability risks.”
This article was published on the ISPE Pharmaceutical Engineering Magazine on April, 2016.
For the full article, please go here.
The article has 5 sections:
- QbD-based biowaiver
- BCS
- Risk assessment in BCS
- Design Space
- Process Analytical Technology (PAT)
To take a quick peek, here are some figures from the article.
Figure 1 shows the FDA and ICH’s progress on developing the concept of biowaiver (In vivo bioequivalence) and how it reached QbD.

The reference documents corresponding to each step are linked below for your convenience.
- SUPAC Immediate Release Solid Oral Dosage Form (SUPAC Immediate Release (Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation)
- SUPAC Modified Release Solid Oral Dosage Form
- IVIVC Extended Release Oral Dosage Forms (Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations)
- Dissolution Testing of Immediate Release Solid Oral Dosage Forms
- FDA’s Experience on IVIVC-New Drug Products – Suarez
- Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on Biopharmaceutics Classificaiton System
- In Vitro-In Vivo Correlation – Linking Drug Release to Clinical Performance – Qiu
- EMEA Guideline on the Investigation of Bioequivalence
- QbD- ICH 8 R2
Figure 2 is a fishbone (Ishikawa) diagram for bioavailability risk for oral dosage forms.
Here, risk sources are categorized into formulation, physiological factors, process and stability.
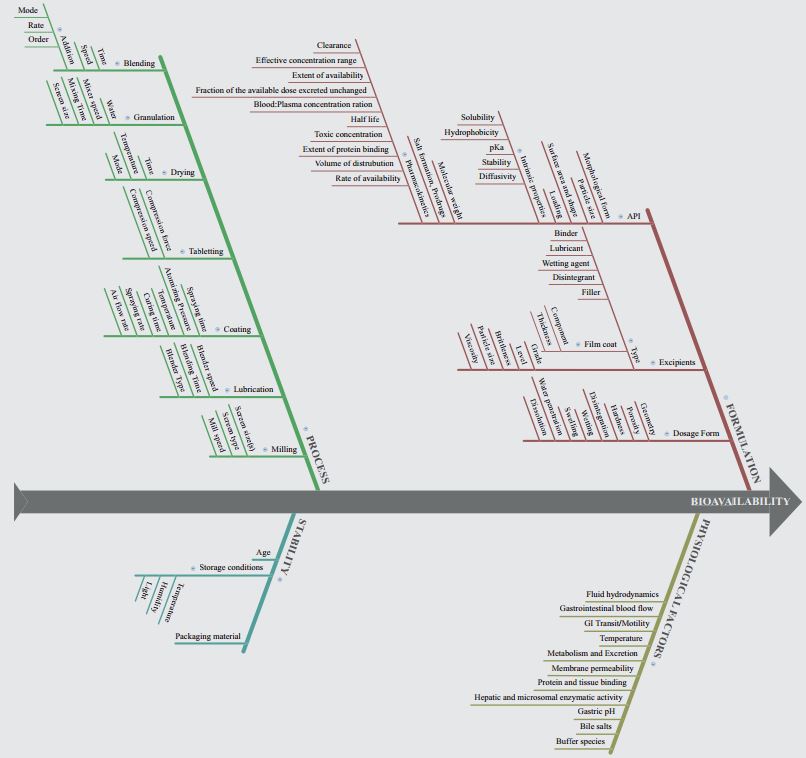
Under each of these categories, we can divide them into sub-categories.
For formulation,
- Dosage Form
- Geometry
- Porosity
- Hardness
- Disintegration
- Wetting
- Swelling
- Water Penetration
- Dissolution
- Excipients
- Type
- Film Coat (Component, Thickness)
- Filler
- Disintegrant
- Wetting Agent
- Lubricant
- Grade
- Level
- Bitterness
- Particle size
- Viscosity
- Type
- API
- Morphological Form
- Particle Size
- Surface area and shape
- Loading
- Intrinsic properties (Diffusivity, Stability, pKa, Hydrophobicity, Solubility)
- Molecular Weight
- Salt formation, Prodrugs
- Pharmacokinetics (Rate of availability, Volume of distribution, Extent of protein binding, Toxic Concentration, Half life, Blood Plasma concentration Ration, Fraction of the available dose excreted unchanged, Extent of availability, Effective concentration range, Clearance)
Physiological factors are:
- Fluid hydrodynamics
- Gastrointestinal Blood flow
- GI Transit/Motility
- Temperature
- Metabolism and Excretion
- Membrane Permeability
- Protein and tissue binding
- Hepatic and microsomal enzymatic activity
- Gastric PH
- Bile salts
- Buffer species
Stability factors are:
- Age
- Storage conditions (Temperature, Humidity, Light)
- Packaging material
And finally, process parameters are:
- Blending
- Time
- Speed
- Addition (Order, Rate, Mode)
- Granulation
- Water
- Mixer Speed
- Mixing time
- Screen size
- Drying
- Time
- Temperature
- Mode
- Tabletting
- Compression force
- Compression speed
- Coating
- Spraying time
- Atomizing Pressure
- Temperature
- Curing time
- Spraying rate
- Air flow rate
- Lubrication
- Blender speed
- Blending time
- Blender type
- Milling
- Screen size (s)
- Screen type
- Mill speed
With tool such as, Lean QbD, you can link these variables into one model for risk assessment.
Lastly, the table below proposes a ranking criteria which links risk with BCS class. This is an area where every development team needs to develop their own criteria and ranking system.

Conclusion
In vivo equivalency is one of the major challenges and also the biggest risk in generic solid dosage development, since its the basis of clinical efficiency of the dosage form. Many valuable studies have been performed, including IVIVC. FDA has also started BioRAM program in 2015, in order to link Biopharmaceutics and QbD.
Thus at this point it’s crucial to use QbD tools and perspective in order to achieve deeper understanding with risk analysis, more mechanistic simulations, and also new PAT tools to investigate the relationship between in vitro and in vivo behaviours.
Mehtap Saydam is a Lean QbD user and contributes regularly to QbDWorks and our Quality-by-Design Linked-In Group. Using tools such as Lean QbD to link CPP, CQA to QTPP will help the team through this process.
Use In Vitro Testing, BCS (Biopharmaceutical Classification Systems), and QbD Risk Assessment to achieve clinical safety/efficacy in QbD.
Here’s the link to read the full ISPE article (need a member access)
Thank you Sun, for giving place to this study here, with a quite interesting summation.
In vivo equivalency is one of the major challenges and also the biggest risk in generic solid dosage development, since its the basis of clinical efficiency of the dosage form. Many valuable studies have been performed, including IVIVC. FDA has also started BioRAM program in 2015, inorder to link Biopharmaceutics and QbD (http://www.aaps.org/uploadedfiles/content/sections_and_groups/focus_groups/selenfdaperspectivesjan2011.pdf).
Thus at this point it’s crucial to use QbD tools and perspective in order to achieve deeper understanding with risk analysis, more mechanistic simulations, and also new PAT tools to investigate the relationship between in vitro and in vivo behaviours.
Thank you Mehtap as always for sharing your knowledge and experience. Edited to reflect this point. Thanks again!